Abstract
Acute myeloid leukemia (AML) is sustained by a population of cancer stem cells (CSCs or cancer-initiating cell). The mechanisms underlying switches from CSCs to non-CSCs in vivo remain to be understood. We address this issue in AML from the aspect of epigenetics using genome-wide screening for DNA methylation and selected histone modifications. We found no major differences in DNA methylation, especially in promoter CpG islands, between CSCs and non-CSCs. By contrast, we found thousands of genes that change H3K4me3 and/or H3K27me3 status between stem and progenitor cells as well as between progenitor and mature cells. Stem cell related pathways and proliferation or metabolism related pathways characterize genes differentially enriched for H3K4me3/H3K27me3 in stem and progenitor populations. Bivalent genes in stem cells are more plastic during differentiation and are more likely to lose H3K4me3 than to lose H3K27me3, consistent with increasingly closed chromatin state with differentiation. Our data indicates that histone modifications but not promoter DNA methylation are involved in switches from CSCs to non-CSCs in AML.
Introduction
Acute myeloid leukemia (AML) is sustained by a population of cancer stem cells (CSCs or cancer-initiating cell) based on the fact that leukemic engraftment could only be initiated from CD34+CD38- fractions.Citation1,Citation2 The mechanisms underlying switches from CSCs to non-CSCs remain to be understood. Two recent studies shed some light on the plasticity of CSC tumor cells. In their study on melanoma, Morrison and his colleaguesCitation3,Citation4 showed that tumors arising both from CD133− cells and from CD133+ cells sorted from an original melanoma re-establish the original ratios of CD133− and CD133+ cells. This experiment indicates that individual cancer cells can recapitulate the marker heterogeneity of the tumors from which they derive, suggesting plasticity of CSCs. More recently, Chaffer et al. described spontaneous conversion of neoplastic non-stem cells to a stem-like state.Citation5 However, the mechanisms underlying the de novo generation of CSCs from non-CSCs in vivo remained to be understood.
Epigenetic regulation of gene expression is mediated in part by DNA methylation and posttranslational modifications such as histone modifications to regulate chromatin structure.Citation6-Citation9 Various observations have suggested that chromatin undergoes remarkable alterations and plays an important role in normal stem cell development.Citation10-Citation12 Furthermore, recent observations suggested that epigenetic-based mechanisms play a key role also in controlling hematopoietic stem cell self-renewal or differentiation.Citation13,Citation14 Tumor development also involves epigenetic changes such as hypomethylation of DNA and hypoacetylation of chromatin, as well as gene specific hypomethylation and hypermethylation.Citation15-Citation18
Emerging data point to a key role for epigenetic regulation in CSCs. Roesch et al. reported differential expression of the H3K4 demethylase JARID1B in subpopulations of human melanoma cells in culture.Citation19 JARID1B+ cells cycled more slowly than JARID1B− cells, but they also generated more progeny and were more tumorigenic, thereby implying that histone methylation patterns determine the CSC state. Epigenetic changes are likely to be a mechanism for distinction of CSCs and non-CSCs since genetic differences have not been found between them. However, little is known about differences in overall chromatin structure between CSCs and non-CSCs. To obtain a better understanding of the epigenomic landscapes in stem and progenitor cells in AML, we used genome-wide analyses for DNA methylation and two key histone modifications, H3K4me3 and H3K27me3. Our data indicates histone modifications but not promoter DNA methylation are involved in switches from CSCs to non-CSCs in AML.
Results
DNA methylation analysis
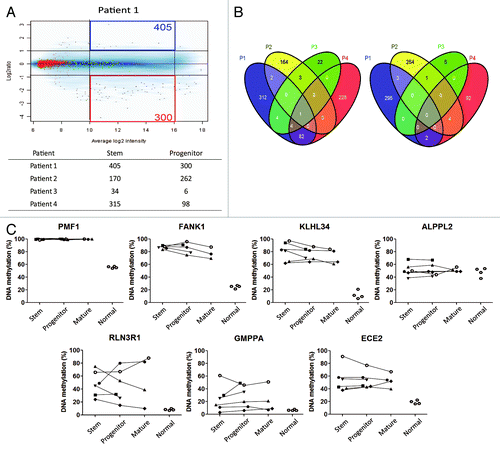
First, we studied the methylation status of five stem cell-related genes as well as four genes known to be differentially methylated in AMLCitation20 (). We found no consistent differences among stem, progenitor, and mature cells in any of the genes analyzed in 6 patients. For example, OCT4 was hypermethylated compared with normal in all fractions analyzed, c-MYC was unmethylated in all and CDH13, when hypermethylated, was equally increased in all fractions. We then proceeded with genome-wide DNA methylation analysis using methylated CpG island amplification microarray (MCAM)Citation21 in 4 patients. Overall, there were few probes which showed > 2 fold signal intensity difference either in stem or progenitor cells (), ranging from 34 to 405 (0.08% to 1%) for probes methylated in stem cells and 6 to 300 (0.01% to 0.7%) for probes methylated in progenitor cells. Very few probes were differentially methylated in ≥ 3 out of 4 patients (5 probes) (). We then looked at probes differentially methylated in at least 2 out of 4 patients. Only 101 (0.2%) probes were differentially methylated (91 probes in stem cells and 10 probes in progenitor cells). Since the specificity of MCAM is known to be around 96%,Citation21 4% of probes are likely to be false positives. We therefore tried to confirm whether those few probes are true positives. We performed quantitative bisulfite-pyrosequencing for 7 individual loci in 6 patients. All tested loci showed no consistent differences by quantitative bisulfite-pyrosequencing () and are therefore false positives. Thus, we found no systematic differences in promoter DNA methylation between AML CSCs and progenitor cells.
Figure 1. DNA methylation level of stem, progenitor, and mature cells for (A) stem cell-related genes and (B) differentially methylated genes in AML by bisulfite-pyrosequencing. Each symbol represents a different patient and lines link results of different cell fractions in the same patients. Normal corresponds to unfractionated peripheral blood cell DNA in four separate control individuals.
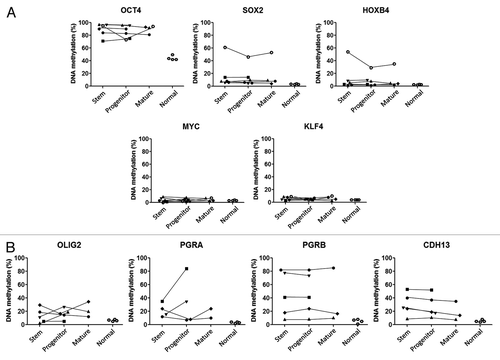
Figure 2. (A) Methylated CpG island amplificationmicroarray analyses (MCAM) of AML cell fractions. A representative MCAM for stem vs progenitor cells from an AML patient (Patient 1). Differentially methylated probes are defined as having average log2 intensity with intermediate signal and > 2 fold higher in stem (blue) or progenitor (red). The numbers of each differentially methylated probe are shown in the corners. Below the graph is a summary of the number of differentially methylated probes (B) Venn diagram for the probes methylated across patients in stem (left) and progenitor (right) cells. (C) DNA methylation level by bisulfite-pyrosequencing for the genes which showed more methylation in AML stem (upper) and in progenitor (lower) cells in MCAM analysis. Each symbol represents a different patient and lines link results of different cell fractions in the same patients. Normal corresponds to unfractionated peripheral blood cell DNA in four separate control individuals.
Chromatin immunoprecipitation-sequencing (ChIP-seq)
We next studied histone modifications by chromatin immunoprecipitation and deep sequencing (ChIP-seq) on 2 patients studied individually and one pool of 3 patients. For the pooled sample (Patients 7–9; P7–9) from three AML patients (total > 1 × 106 in each population; 0.3 × 106 in each ChIP), we used the standard method for ChIP-seq. We generated 6.7–23 million unique usable tags (quality filtered and aligned to the human genome) for each of H3K4me3 (K4), H3K27me3 (K27), and H3 as a control (Table S3). To enable ChIP-seq analysis from small numbers of cells (2 × 105 in each population; 0.6 × 105 in each ChIP) isolated from individual patients (Patient 10 and Patient 11; P10 and P11), we optimized ChIP and library preparation. We confirmed that ChIP with small numbers of cells worked as expected and library preparation did not generate PCR bias by comparing samples before and after amplification for positive and negative control genes by quantitative PCR (Fig. S1). With these methods, we generated 6.4–26 million unique usable tags for each ChIP sample (Table S3). To investigate cell type-specific histone modification patterns, we employed a spatial clustering approach for the identification of ChIP-enriched regions using the SICER algorithm.Citation22 Peaks generated from these tags revealed that K4 is concentrated at promoter regions whereas K27 is broadly distributed from transcription start sites (TSS) to transcription end sites (TES) covering the entire genes (Fig. S2). These findings are similar to previous reports,Citation23 confirming the reliability and accuracy of the methods and data analyses. This pattern of localized K4 peaks and broad K27 peaks was very similar between stem and progenitor cells.
Histone modification patterns and gene expression
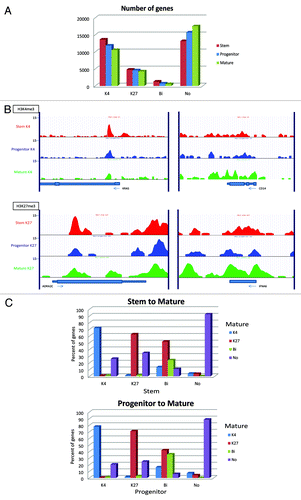
By ChIP-seq analysis of the sample P10, we identified K4 peaks at the promoters of 14,891 and 12,562 genes in stem and progenitor cells, respectively, while K27 peaks were found in 6,050 and 5,294 genes, respectively (Fig. S2). There were relatively few K4 or K27 peaks outside of gene. Because K4 and K27 tend to be inversely correlated in their gene expression,Citation24 we categorized genes into K4 only, K27 only, bivalent, and bi-negative. Using publicly available AML stem and progenitor expression patterns (accession number GSE24006),Citation25 we categorized genes depending on their expression into high, intermediate, and low in stem and progenitor cells, and correlated these with K4 and K27 peaks (). We found that high expression genes had clearly higher K4 enrichment in stem cell and progenitor cell data sets. The reverse was found for K27. Whereas K4 enrichment was found to be concentrated at the TSS region, K27 enrichment was distributed broadly from the TSS to TES. Similar results were found using a separate gene expression data set (data not shown).Citation26 We then calculated average expression z-scores in the groups of genes with K4 only, K27 only, bivalent, and bi-negative in stem and progenitor cells. In both data sets, we found that K4 only genes have the highest expression, while K27 only genes, bivalent, and bi-negative genes were equally repressed (). Overall, progenitor cells had somewhat fewer K4 only genes, K27 only genes, and bivalent genes whereas the number of bi-negative genes was increased (). Similar data were seen for P11 and P7–9 (Fig. S3).
Figure 3. (A) Histone mark enrichment by gene expression patterns in P10. Gene expression patterns were divided into high (top 2,000 genes), intermediate (medium 2000 genes), and low (bottom 2000 genes). (B) Categories of promoter histone marks (K4 only, K27 only, bivalent and none) and associated gene expression levels. (C) Numbers of the genes with respective histone status. Gene expression data was taken from GEO data set GSE24006.

Differences between AML stem and progenitor cells
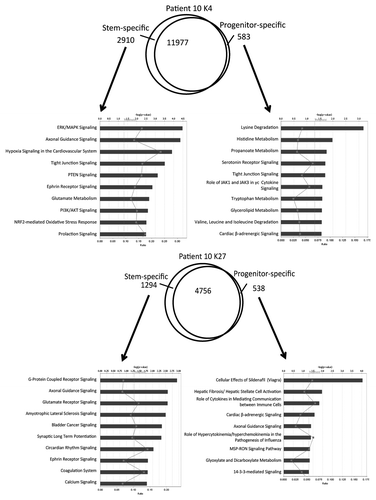
From P10, we found 3,493 differentially K4-enriched genes in stem and progenitor cells (2910 genes in stem cells and 583 genes in progenitor cells, respectively), which correspond to only 77.5% conservation of K4 status between the two cellular compartments (). In pathway analysis of the K4 data, “ERK/MAPK signaling” and “hypoxia signaling” were found to be enriched in the stem cell population whereas several metabolism pathways such as “lysine degradation” were enriched in the progenitor cell population. We also found 1,832 differentially K27-enriched genes (1,294 genes in stem cells and 538 genes in progenitor cells) (), which correspond to 70% conservation in K27 status in stem and progenitor cells. When we included both K4 variable and K27 variable genes in pathway analysis, we found enrichment for differentiation and metabolism pathways as well (Fig. S4). Qualitatively similar differences in numbers of differentially K4-or K27-enriched were found in P11 and P7–9 (Fig. S5). We next looked for commonly altered pathways in the 3 ChIP-seq data sets. Among the observed pathways were “ephrin receptor signaling” for K4 (P10 and P7–9) in stem cells and “hepatic fibrosis/hepatic stellate cell activation signaling” for K27 (P10, P11 and P7–9) in progenitor cells. This was also the case for an analysis that included both K4 and K27 variables. When we focused on common differentially marked gene in P10, P11, and P7–9, very few overlapped completely, highlighting patient-to-patient heterogeneity (Fig. S6). To illustrate this further, we performed unsupervised hierarchical clustering on the K4 and K27 data sets (Fig. S7A). While K4 and K27 clustered separately, within each mark, the stem and progenitor profiles clustered intra-patient rather than by cell subtype. Similar data were seen for gene expression clustering (Fig. S7B).
Figure 4. Pathway analysis for genes enriched in K4 (upper) or K27 (lower) in P10. The Venn diagrams show overlap in genes marked by K4 or K27 in their promoters. Ingenuity pathway analysis was performed with the exclusively enriched genes in stem or progenitor cells. Bars show p values in –log (p value) for each pathway. Rectangles on each bar show ratio calculated by the number of molecules in a given pathway divided by total number of molecules that make up that pathway. Pathways are listed in order of a rank from the most associated pathway.
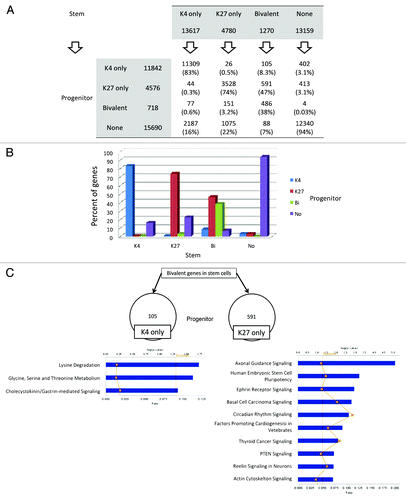
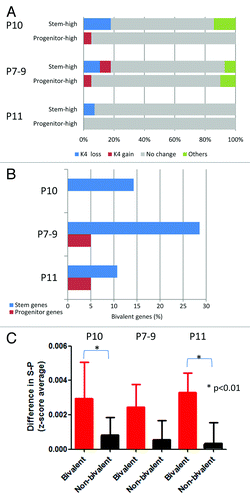
We then examined specific switches in chromatin status from stem to progenitor cells (; Fig. S8). In P10, most of the genes with K4, K27 or bi-negative status in stem cells did not change histone status in progenitor cells (74% to 94%). We also found that full switch events (K4 in stem cell to K27 in progenitor cells or the reverse case) are very rare (< 100 genes). In contrast, bivalent genes were the least conserved between stem and progenitor cells (38%). Interestingly, bivalent genes were more likely to lose K4 than lose K27 upon differentiation from stem to progenitor cells. For example, out of the 1,270 bivalent genes in stem cells in Patient 10, 591 (47%) genes lost K4 marks, 105 (8.3%) genes lost K27 marks, and 88 (7%) lost both K4 and K27 marks after differentiation into progenitor cells. Pathway analysis revealed that neuronal differentiation-related pathways such as “axonal guidance signaling” and “ephrin receptor signaling” are enriched among genes that lost K4 upon differentiation. In contrast to P10, bivalent genes were mostly conserved in P7–9 (82%) and P11 (79%). Nevertheless, bivalent genes both in P7–9 and P11 were more likely to lose K4 than lose K27 upon differentiation from stem to progenitor cells (3 fold more likely in P7–9 and 7 fold more likely in P11). Pathways such as G-protein coupled receptor signaling and synaptic long-term depression signaling are found to be enriched both in P7–9 and P11.
Figure 5. Histone switches in AML stem vs. progenitor cells. (A) Number of genes in each histone category in stem and progenitor AML cells. Arrows point to the histone status in progenitor cells by baseline histone status in stem cells. Thus, of 13617 K4 only promoters in stem cells, 83% remain K4 only in progenitor cells and 17% switch to other histone status. (B) Graphical representation of data shown in (A). (C) Pathway analysis for the genes that are bivalent in stem cells and switch to K4 only or K27 only in progenitor cells.
To correlate histone modification patterns in stem and progenitor cells with gene expression, we studied genes previously found to be differentially expressed in AML stem/progenitor cells. These were 28 stem cell specific genes and 20 progenitor cell specific genes.Citation25 Since we found that K4 was the most reliable marks of gene expression), we focused on loss and gain of K4 enrichment upon differentiation. In P10 AML cells, we found that 5 out of 28 (18%) stem cell-expressed genes had only K4 marks in stem cells that were lost in progenitor cells compared with only 1 out of 20 progenitor cell-expressed gene (5%) (). Thus the majority of gene expression changes detected in stem vs. progenitor cells cannot be attributed to chromatin changes. Similar data were found in P7–9 and P11. When factoring in K27, we found that stem cell-expressed genes were enriched in the bivalent status in stem and/or progenitor cells compared with progenitor cell-expressed genes (). In other words, bivalent genes appeared to contribute significantly to the factor of genes that have expression upon stem cell differentiation. To study this issue further, we examined the difference in expression between stem and progenitor cell fractions (stem – progenitor). This difference was significantly higher for bivalent genes in stem cells compared with non-bivalent genes in 2 out of 3 patients analyzed (p < 0.01 for P10 and P11) ().
Figure 6. Relation between switches in histone status and differential expression in AML. (A) K4 switches for genes that show high expression in stem cells (stem-high) or in progenitor cells (progenitor-high). In each patient (P10, P7–9, and P11), most genes show no histone switches (gray), but a significant minority show consistent changes (K4 switches tracking with expression switches). (B) Percent of bivalent promoters among genes that show differential expression in stem or progenitor cells. (C) Bivalent genes in stem cells (red) show preferential loss of expression compared with non-bivalent genes (black).
Chromatin changes in the differentiated cells
In the patient with high blast percentage (P10), we were able to obtain enough fully differentiated (CD34-) cells for ChIP-seq analysis. We found the number of genes with K4 only, K27 only, and bivalent status decreased with differentiation from stem cells to progenitor cells and mature cells (), with a concomitant increase in the number of bi-negative genes. This was most striking for bivalent genes that decreased to less than half of those in stem cells (1270 genes and 528 genes in stem and mature cells, respectively). Next, we sought to find genes that are differentially enriched with K4 and K27. shows examples of K4-enrichment for a stem-cell gene (KRAS) and a differentiation marker (CD14) and K27-enrichment of a stem-cell gene (ADRA2C) and a mature-cell gene (IFNA6). Interestingly, we could find more K4-enriched genes that are overlapped only between stem and progenitor cells than those only between stem and mature cells (2432 and 864 genes, respectively) (Fig. S9). This is also true for K27-enriched genes (1042 and 316 genes, respectively) , suggesting a greater similarity in chromatin status between stem and progenitor cells. Pathway analysis for the genes enriched only in each population revealed similar pathways as found in the analysis between stem and progenitor cells. In addition, pathways related to inflammation and immune system in macrophages are enriched in mature cells. Histone status is less conserved between stem and mature cells than those between stem and progenitor cells (72% vs. 83% for K4 only, 63% vs. 74% for K27 only, 24% vs. 38% for bivalent, and 92% v.s 94% for bi-negative genes, respectively) ( and ). Nevertheless, we still could see the trend that bivalent genes are more likely to lose K4 than K27 upon differentiation from stem to mature cells (52% vs. 14%) as well as from progenitor to mature cells (42% vs. 16%).
Figure 7. Chromatin changes from stem to progenitor to the most differentiated cells (mature cells). (A) Comparison of numbers of promoter genes with respective histone status. (B) Representative histone modification patterns of K4-enrichment for a stem-cell gene (KRAS) and a differentiation marker (CD14) and K27-enrichment of a stem-cell gene (ADRA2C) and a mature-cell gene (IFNA6). (C) K4/K27 mark change upon differentiation in Patient 10. Percentages of each histone status are shown in in bar graphs for differentiation from stem to mature cells (upper) and from progenitor to mature cells (lower).
Discussion
In this paper, we compared the epigenome of AML stem and progenitor cells and found no systematic differences in DNA methylation, but substantial differences in chromatin status. The lack of differential methylation was confirmed for 5 stem cell-related genes, 4 genes known to be differentially methylated in AML, as well as genome wide analysis (by MCAM). One of reasons to explain this is a limited coverage of MCAM, which mainly focused on promoter DNA methylation. Although we found some differentially methylated probes also at non-promoter regions in MCAM, it does not seem to have any preference compared with those at promoter regions (data not shown). However, we cannot completely exclude the possibility of differences in DNA methylation outside promoter regions. It has been suggested recently that DNA methylation in intergenic regions might play an important role in differentiation.Citation27 Next generation sequencing technologies might enable the clarification of this. Alternatively, DNA methylation could be uninvolved in the switches from CSCs to non-CSCs and tumor hierarchy in AML. According to genomic studies, most developmental transcriptional factors are not regulated by DNA methylation.Citation28 Furthermore, current researches support a plastic CSC model whereby both populations can switch their status when needed. DNA methylation provides a mechanism for long-term epigenetic memoryCitation29 and thus may not be appropriate to regulate potentially reversible differentiation states in cancer. Histone modifications clearly are essential mediators of stem cell plasticity in normal states. For example, genes repressed by Polycomb (PcGs) and marked by H3K27me3 in ES cells retain the potential to become activated after lineage commitment, implying that the epigenetic marks need to be reversible.Citation30-Citation32 This is also true for genes specifying the neuronal cell fate, which are repressed by PcGs in undifferentiated cells and become activated through displacement of PcGs in response to the induction of differentiation.Citation31
We studied two key histone modifications, H3K4me3 and H3K27me3, since several papers have shown their importance in regulation of development and differentiation.Citation23,Citation33 Our data suggest that histone modifications also mediate stem cell to differentiated cell transition in cancer. In general we found as expected that K4 marks are concentrated at TSS whereas K27 marks are distributed broadly.Citation34,Citation35 The genes enriched with K4 and/or K27 marks showed different levels of gene expression both in stem and progenitor cells; K4 only genes showed highest expression whereas K27 only genes showed lowest expression. Bi-negative genes showed almost same level as bivalent genes but slightly higher. Bivalent genes were generally repressed but not as much as K27 only gene, reflecting status as “poised” for expression. Interestingly, almost 40% of genes were “bi-negative,” lacking both K4 and K27 marks. These are rare in ES cells and thus are characteristic of more lineage-committed cells. These genes had slightly higher expression than bivalent genes, and are clearly not a measurement artifact since they were quite conserved between stem and progenitor cells. Presumably, these promoters are marked by alternate histone modifications such as H3K9me2. These findings are similar to what has been reported so far in hematopoietic stem cells,Citation36 suggesting that histone modifications regulate gene expression in individual cell population also in AML cells.
There were substantial differences in histone marks between stem and progenitor cells. Although we found few differentially marked genes that are common to all patients’ samples analyzed, there were several common functional pathways affected. Among the observed pathways were “ephrin receptor signaling” for K4 (P10 and P7–9) and “hepatic fibrosis/hepatic stellate cell activation signaling” for K27 (P10, P11, and P7–9), implying common regulatory mechanisms at a pathway level to orchestrate differentiation from stem to progenitor cells. Both of these signaling pathways are also known to function in neural and intestinal stem cells,Citation37-Citation39 implying common features of stem cells between different tissues, and even different conditions, that is, normal and tumor. When factoring in the most differentiated cells in P10, we found pathways related to the function of differentiated myeloid cells, suggesting the importance of histone modifications for further terminal differentiation. On the other hand, only a small number of differentially expressed genes in stem or progenitor cells could be explained by differences in histone status. Similar data were described for breast cell differentiation.Citation33 These findings are consistent with a recent hypothesis that histone modifications indicate, instead of tight regulation of transcription, fine-tuning of transcription to ensure a certain chromatin state that allows the proper execution of transcriptional control.Citation40 Overall, our results suggest that genome-wide histone modifications are important to determine stem or progenitor cell states in AML.
A bivalent chromatin status described as having both K4/K27 marks at promoters was first seen in embryonic stem cells and thought to keep poised expression in key developmental genes.Citation23 It has been reported in hematopoietic stem cells that bivalent genes lose either K4 or K27 marks after differentiation.Citation41 Although their function in cancer is not yet clear, bivalent genes might also be involved in cancer stem cell plasticity. We found bivalent genes are more likely to lose K4 than lose K27 in progenitor cells (3 to 7 times more likely to lose K4 than lose K27) and mature cells as well in P10. This is similar to normal hematopoietic stem cell differentiation,Citation41 suggesting conserved mechanisms for regulating normal and AML stem states. Interestingly, pathway analysis revealed that neuronal differentiation-related pathways such as “axonal guidance signaling” and “ephrin receptor signaling” are enriched among bivalent genes that lose K4, implying the loss of potential for differentiation into other lineages. Consistent with their histone modifications switches, bivalent genes in stem cells are more likely to be expressed in stem than in progenitor cell, implying that these pathways are regulated by having bivalency in stem cells to retain poised status before they are silenced in progenitor cells by losing the K4 mark to shut down their expression.Citation23 Taken together, it seems that bivalent genes play an important role in regulating differentiation and that this is a general phenomenon from embryonic stem cells to hematopoietic stem cells and AML stem cells.
Altogether, our data indicates that histone modifications but not promoter DNA methylation are involved in switches from CSCs to non-CSCs in AML. In this regard, analyses of other histone modifications such as H3K9me2, H3K9me3 or histone marks of enhancers might also help to understand more precise mechanisms of differences between stem and progenitor cells. It has been reported that mammalian cells acquire large regions of H3K9me2 during differentiation, which affect at least 30% of the genomeCitation42 and H3K9me3 expands in fibroblasts upon differentiation.Citation10 Bivalent domains in many cell types have a high degree of association with additional histone modifications to form combinatorial marked chromatin domains.Citation41,Citation43 Furthermore, regulation by chromatin states could also involve upstream enhancers, locus control regions, and transcription factors.Citation44-Citation46 Still, the fact that chromatin switches occur prominently in the transition from CSCs to non-CSCs has implications for therapeutic targeting of epigenetic mechanisms in AML.
Materials and Methods
Patients and cells
Peripheral blood samples in AML patients were separated to obtain stem cells (CD34+CD38-), progenitor cells (CD34+CD38+), and mature cells (CD34-) by magnetic cell sorting (MACS®, Myltenyi Biotec). A summary of sample usage for DNA methylation and histone modification analyses is listed in Table S1. The institutional review board at MD Anderson approved all protocols, and all patients gave informed consent for the collection of residual tissues as per institutional guidelines and in accordance with the Declaration of Helsinki.
DNA methylation analysis
DNA methylation level of 5 stem cell-related genes (OCT4, SOX2, MYC, HOXB4 and KLF4) was analyzed for their promoters using a quantitative bisulfite-pyrosequencing assay.Citation47 Four promoters of genes (OLIG2, PGRA, PGRB and CDH13) known to be methylated in AMLCitation20 were analyzed as well. Primer sequences are listed in Table S2.
For genome-wide analysis, we performed methylated CpG island amplification microarray (MCAM) as previously reported.Citation21,Citation48 In brief, MCA amplicons of stem cells and progenitor cells were Cy5- and Cy3-labeled, respectively, and cohybridized to a custom 4 × 44 k oligo array (GEO accession number GPL9886, Agilent Technologies) where 7202 promoter CpG islands, 1348 non-promoter CpG islands, and 632 non-CpG island promoter methylation can be detected.
Chromatin Immunoprecipitation-sequencing (ChIP-seq)
For histone modification analysis, we used ChIP-Seq for stem cell and progenitor cell populations for H3K4me3 and H3K27me3, which are known to be markers for activated and silenced genes, respectively. To overcome technical challenges in the use of small numbers of cells for ChIP-seq, we modified ChIP protocols and library preparation for sequencing. In brief, we were able to perform ChIP-Seq experiments for histone modifications from < 1 ng ChIP-ed DNA as a starting material. Following cell purification, 2 × 105 cells were washed with PBS, then immediately cross-linked with formaldehyde (final concentration was 0.5%) at room temperature for 8 min. The reaction was stopped by incubation with glycin (1.25 M) for 5 min., and cells were washed two times with 1ml PBS+BSA (5mg/ml). Cells were re-suspended in 200 μl SDS lysis buffer (1% SDS, 10 mM EDTA, 50 mM TRIS-HCl pH 8.1, and protease inhibitors: Complete mini, Roche) and incubated on ice for 10 min. Cell lysates were then sonicated using a Bioruptor (Diagenode) for 30 sec at the maximum setting 15 times with 30 sec intervals. The sonicated lysates were centrifuged for 10 min., the supernatant was 10× diluted with 1800 μl of dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 167 mM NaCl, 16.7 mM TRIS-HCl pH 8.1), then transferred to 30 μl pre-washed protein G magnetic beads (Invitrogen), and incubated at 4°C overnight. One μg of anti-H3K27me3 antibody (07-449, Millipore) or 0.2 μg of anti-H3K4me3 antibody (ab8580, Abcam), were pre-incubated with 10 μl of protein G magnetic beads at 4°C overnight before immunoprecipitation. 600 μl of the pre-cleared sheared chromatin was incubated with the antibody-coated protein G magnetic beads at 4°C for overnight. The immunoprecipitates were washed (using magnetic stand) six times with 1ml wash buffer (0.1% SDS, 1% Triton X-100, 1 mM EDTA, 0.5 mM EFTA, 10 mM TRIS-HCl pH 7.5, 140 mM NaCl, 0.1% Na-deoxycholate). Tubes were incubated on rotator for 5 min at 4°C between every wash. Then they were washed once with 1 ml TE. Samples were re-suspended in 100 μl of elution buffer (10% SDS and 0.5 M EDTA, 1 M TRIS-HCl pH 8.0) and were incubated at 65°C for 2 h to reverse crosslinking, followed by RNase A and proteinase K treatment for 30 min. The recovered DNA was purified using PCR purification kit (MinElute PCR Purification Kit, QIAGEN, Valencia, CA). Part of ChIP-ed DNA was tested by qPCR for GAPDH and RARB as controls for K4 and K27, respectively, and the remaining DNA was end-repaired and purified using a PCR purification kit (QIAGEN) and resuspended in 34 μl of TB buffer. “A” base was added to the 3′-ends by Klenow. DNA was purified using the PCR purification kit and resuspended in 10 μl of TB buffer and 1:100 diluted Adaptor oligo mix (ChIP-Seq Sample Preparation Kit, Illumina), ligated using T4 DNA ligase HC (Enzymatics). Fragments ranging from 250–300 bp were isolated from 2% agarose gels followed by purification using the PCR purification kit and subjected to PCR amplification (18 cycles) using specific primers provided in the kit. Amplified DNA was purified using magnetic beads (Agencourt Ampure XP, Beckman Coulter), and re-tested by qPCR for GAPDH and RARB to compare with the results obtained from purified DNA before amplification.
Bioinformatics
Sequenced DNA tags were mapped to human genome hg18 using ELAND (Illumina) and uniquely mapped tags were kept. To avoid the PCR bias of multiple tags that were mapped to the same genomic location, only one copy was kept for further analysis. SICER (version 1.03)Citation22 was used to detect peaks and enriched domains. The window size was set as 200 bp as default. The gap size was determined as recommended by Zang et al.,Citation22 or at most 2 kb, since the performance worsens as the gap size increases more than 10 times the window size. Following Wang et al.,Citation49 E-value was set at FDR ≤ 5%, which was estimated as E-value (the expected number of significant domains under the random background) divided by the number of identified candidate domains. The FDR cutoff was set as 5% to further filter out the candidate domains by comparing to the control. Each peak/domain was assigned to the gene that has the closest TSS to the peak/domain. Then the peak was classified by its location to the gene: upstream (-5 k to -1 k from TSS), promoter (-1 k to +0.5 k from TSS), exon, intron, TES (-0.5 k to + 1k from TES) and downstream (+1 k to +5 k from TES). The gene list used to annotate the peaks is the RefSeq gene list downloaded from UCSC genome browser (http://genome.ucsc.edu/) at April 2010. When the promoter region (-1 kb to +0.5 kb from TSS) of a gene was overlapped with peaks, the gene was defined as marked by H3K4me3 or H3K27me3. For gene expression analysis, 5 kb upstream of TSS and 5 kb downstream of TES were subdivided into 1 kb bins and the gene body was subdivided evenly into 20 bins for each gene. In each bin, tags were summed over and normalized by the length of the bin and the total number of tags in the genome. The normalized number of tags of H3K4me3/H3K27me3 in each corresponding bin was then subtracted by that of H3 and averaged over three groups of genes according to their expression levels (highest expression, intermediate expression and lowest expression). Each group had 2,000 genes. For landscapes of the data, each tag was extended 200 bp to its 3′ end. Then, the number of overlapped tags in each genome position was rescaled to normalize the total number of tags to 10 M and averaged over 10 bp resolution. The averaged values were displayed using UCSC genome browser.
Gene expression data analysis
Publically available gene expression data was used to obtain gene expression levels in stem and progenitor cells (GEO accession number GSE24006).Citation25 We calculated z-score to standardize expression levels of each gene on the arrays. We then computed average z-score in AML in stem vs. progenitor cells. We also studied separately 28 stem and 20 progenitor-specific genes previously identified based on this data set.Citation25
Additional material
Download Zip (1.5 MB)Acknowledgments
The authors thank the patients who have contributed to our understanding of these disorders. This work was supported by National Institutes of Health grants CA100632, CA121104 and CA046939 and supported by a Stand Up to Cancer grant from the American Association for Cancer Research. J.P.I. is an American Cancer Society Clinical Research professor supported by a generous gift from the F.M. Kirby Foundation.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/epigenetics/article/23243
References
- Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med 1997; 3:730 - 7; http://dx.doi.org/10.1038/nm0797-730; PMID: 9212098
- Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994; 367:645 - 8; http://dx.doi.org/10.1038/367645a0; PMID: 7509044
- Quintana E, Shackleton M, Sabel MS, Fullen DR, Johnson TM, Morrison SJ. Efficient tumour formation by single human melanoma cells. Nature 2008; 456:593 - 8; http://dx.doi.org/10.1038/nature07567; PMID: 19052619
- Shackleton M, Quintana E, Fearon ER, Morrison SJ. Heterogeneity in cancer: cancer stem cells versus clonal evolution. Cell 2009; 138:822 - 9; http://dx.doi.org/10.1016/j.cell.2009.08.017; PMID: 19737509
- Chaffer CL, Brueckmann I, Scheel C, Kaestli AJ, Wiggins PA, Rodrigues LO, et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc Natl Acad Sci U S A 2011; 108:7950 - 5; http://dx.doi.org/10.1073/pnas.1102454108; PMID: 21498687
- Jenuwein T, Allis CD. Translating the histone code. Science 2001; 293:1074 - 80; http://dx.doi.org/10.1126/science.1063127; PMID: 11498575
- Margueron R, Trojer P, Reinberg D. The key to development: interpreting the histone code?. Curr Opin Genet Dev 2005; 15:163 - 76; http://dx.doi.org/10.1016/j.gde.2005.01.005; PMID: 15797199
- Bird A. DNA methylation patterns and epigenetic memory. Genes Dev 2002; 16:6 - 21; http://dx.doi.org/10.1101/gad.947102; PMID: 11782440
- Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet 2003; 33:Suppl 245 - 54; http://dx.doi.org/10.1038/ng1089; PMID: 12610534
- Hawkins RD, Hon GC, Lee LK, Ngo Q, Lister R, Pelizzola M, et al. Distinct epigenomic landscapes of pluripotent and lineage-committed human cells. Cell Stem Cell 2010; 6:479 - 91; http://dx.doi.org/10.1016/j.stem.2010.03.018; PMID: 20452322
- Meissner A. Epigenetic modifications in pluripotent and differentiated cells. Nat Biotechnol 2010; 28:1079 - 88; http://dx.doi.org/10.1038/nbt.1684; PMID: 20944600
- Gaspar-Maia A, Alajem A, Meshorer E, Ramalho-Santos M. Open chromatin in pluripotency and reprogramming. Nat Rev Mol Cell Biol 2011; 12:36 - 47; http://dx.doi.org/10.1038/nrm3036; PMID: 21179060
- Akashi K, He X, Chen J, Iwasaki H, Niu C, Steenhard B, et al. Transcriptional accessibility for genes of multiple tissues and hematopoietic lineages is hierarchically controlled during early hematopoiesis. Blood 2003; 101:383 - 9; http://dx.doi.org/10.1182/blood-2002-06-1780; PMID: 12393558
- Zardo G, Cimino G, Nervi C. Epigenetic plasticity of chromatin in embryonic and hematopoietic stem/progenitor cells: therapeutic potential of cell reprogramming. Leukemia 2008; 22:1503 - 18; http://dx.doi.org/10.1038/leu.2008.141; PMID: 18548105
- Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer 2004; 4:143 - 53; http://dx.doi.org/10.1038/nrc1279; PMID: 14732866
- Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet 2002; 3:415 - 28; PMID: 12042769
- Feinberg AP. An epigenetic approach to cancer etiology. Cancer J 2007; 13:70 - 4; http://dx.doi.org/10.1097/PPO.0b013e31803c6e3b; PMID: 17464249
- Jones PA, Baylin SB. The epigenomics of cancer. Cell 2007; 128:683 - 92; http://dx.doi.org/10.1016/j.cell.2007.01.029; PMID: 17320506
- Roesch A, Fukunaga-Kalabis M, Schmidt EC, Zabierowski SE, Brafford PA, Vultur A, et al. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell 2010; 141:583 - 94; http://dx.doi.org/10.1016/j.cell.2010.04.020; PMID: 20478252
- Kroeger H, Jelinek J, Estécio MR, He R, Kondo K, Chung W, et al. Aberrant CpG island methylation in acute myeloid leukemia is accentuated at relapse. Blood 2008; 112:1366 - 73; http://dx.doi.org/10.1182/blood-2007-11-126227; PMID: 18523155
- Estecio MR, Yan PS, Huang TH, Issa JP. Methylated CpG Island Amplification and Microarray (MCAM) for High-Throughput Analysis of DNA Methylation. CSH Protoc 2008; 2008:pdb.prot4974
- Zang C, Schones DE, Zeng C, Cui K, Zhao K, Peng W. A clustering approach for identification of enriched domains from histone modification ChIP-Seq data. Bioinformatics 2009; 25:1952 - 8; http://dx.doi.org/10.1093/bioinformatics/btp340; PMID: 19505939
- Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006; 125:315 - 26; http://dx.doi.org/10.1016/j.cell.2006.02.041; PMID: 16630819
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, et al. High-resolution profiling of histone methylations in the human genome. Cell 2007; 129:823 - 37; http://dx.doi.org/10.1016/j.cell.2007.05.009; PMID: 17512414
- Gentles AJ, Plevritis SK, Majeti R, Alizadeh AA. Association of a leukemic stem cell gene expression signature with clinical outcomes in acute myeloid leukemia. JAMA 2010; 304:2706 - 15; http://dx.doi.org/10.1001/jama.2010.1862; PMID: 21177505
- Gal H, Amariglio N, Trakhtenbrot L, Jacob-Hirsh J, Margalit O, Avigdor A, et al. Gene expression profiles of AML derived stem cells; similarity to hematopoietic stem cells. Leukemia: official journal of the Leukemia Society of America. Leukemia Research Fund, UK 2006; 20:2147 - 54; http://dx.doi.org/10.1038/sj.leu.2404401
- Stadler MB, Murr R, Burger L, Ivanek R, Lienert F, Schöler A, et al. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature 2011; 480:490 - 5; PMID: 22170606
- Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J, Sivachenko A, et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 2008; 454:766 - 70; PMID: 18600261
- Raynal NJ, Si J, Taby RF, Gharibyan V, Ahmed S, Jelinek J, et al. DNA methylation does not stably lock gene expression but instead serves as a molecular mark for gene silencing memory. Cancer Res 2012; 72:1170 - 81; http://dx.doi.org/10.1158/0008-5472.CAN-11-3248; PMID: 22219169
- Boyer LA, Plath K, Zeitlinger J, Brambrink T, Medeiros LA, Lee TI, et al. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature 2006; 441:349 - 53; http://dx.doi.org/10.1038/nature04733; PMID: 16625203
- Bracken AP, Dietrich N, Pasini D, Hansen KH, Helin K. Genome-wide mapping of Polycomb target genes unravels their roles in cell fate transitions. Genes Dev 2006; 20:1123 - 36; http://dx.doi.org/10.1101/gad.381706; PMID: 16618801
- Lee TI, Jenner RG, Boyer LA, Guenther MG, Levine SS, Kumar RM, et al. Control of developmental regulators by Polycomb in human embryonic stem cells. Cell 2006; 125:301 - 13; http://dx.doi.org/10.1016/j.cell.2006.02.043; PMID: 16630818
- Maruyama R, Choudhury S, Kowalczyk A, Bessarabova M, Beresford-Smith B, Conway T, et al. Epigenetic regulation of cell type-specific expression patterns in the human mammary epithelium. PLoS Genet 2011; 7:e1001369; http://dx.doi.org/10.1371/journal.pgen.1001369; PMID: 21533021
- Koch CM, Andrews RM, Flicek P, Dillon SC, Karaöz U, Clelland GK, et al. The landscape of histone modifications across 1% of the human genome in five human cell lines. Genome Res 2007; 17:691 - 707; http://dx.doi.org/10.1101/gr.5704207; PMID: 17567990
- Pauler FM, Sloane MA, Huang R, Regha K, Koerner MV, Tamir I, et al. H3K27me3 forms BLOCs over silent genes and intergenic regions and specifies a histone banding pattern on a mouse autosomal chromosome. Genome Res 2009; 19:221 - 33; http://dx.doi.org/10.1101/gr.080861.108; PMID: 19047520
- Weishaupt H, Sigvardsson M, Attema JL. Epigenetic chromatin states uniquely define the developmental plasticity of murine hematopoietic stem cells. Blood 2010; 115:247 - 56; http://dx.doi.org/10.1182/blood-2009-07-235176; PMID: 19887676
- Jaeger I, Arber C, Risner-Janiczek JR, Kuechler J, Pritzsche D, Chen IC, et al. Temporally controlled modulation of FGF/ERK signaling directs midbrain dopaminergic neural progenitor fate in mouse and human pluripotent stem cells. Development 2011; 138:4363 - 74; http://dx.doi.org/10.1242/dev.066746; PMID: 21880784
- Granero-Moltó F, Myers TJ, Weis JA, Longobardi L, Li T, Yan Y, et al. Mesenchymal stem cells expressing insulin-like growth factor-I (MSCIGF) promote fracture healing and restore new bone formation in Irs1 knockout mice: analyses of MSCIGF autocrine and paracrine regenerative effects. Stem Cells 2011; 29:1537 - 48; http://dx.doi.org/10.1002/stem.697; PMID: 21786367
- Biteau B, Jasper H. EGF signaling regulates the proliferation of intestinal stem cells in Drosophila. Development 2011; 138:1045 - 55; http://dx.doi.org/10.1242/dev.056671; PMID: 21307097
- Kooistra SM, Helin K. Molecular mechanisms and potential functions of histone demethylases. Nat Rev Mol Cell Biol 2012; 13:297 - 311; PMID: 22473470
- Cui K, Zang C, Roh TY, Schones DE, Childs RW, Peng W, et al. Chromatin signatures in multipotent human hematopoietic stem cells indicate the fate of bivalent genes during differentiation. Cell Stem Cell 2009; 4:80 - 93; http://dx.doi.org/10.1016/j.stem.2008.11.011; PMID: 19128795
- Wen B, Wu H, Shinkai Y, Irizarry RA, Feinberg AP. Large histone H3 lysine 9 dimethylated chromatin blocks distinguish differentiated from embryonic stem cells. Nat Genet 2009; 41:246 - 50; http://dx.doi.org/10.1038/ng.297; PMID: 19151716
- Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 2007; 448:553 - 60; http://dx.doi.org/10.1038/nature06008; PMID: 17603471
- Grosveld F. Activation by locus control regions?. Curr Opin Genet Dev 1999; 9:152 - 7; http://dx.doi.org/10.1016/S0959-437X(99)80023-9; PMID: 10322132
- Rosenbauer F, Tenen DG. Transcription factors in myeloid development: balancing differentiation with transformation. Nat Rev Immunol 2007; 7:105 - 17; http://dx.doi.org/10.1038/nri2024; PMID: 17259967
- Rosenfeld MG, Lunyak VV, Glass CK. Sensors and signals: a coactivator/corepressor/epigenetic code for integrating signal-dependent programs of transcriptional response. Genes Dev 2006; 20:1405 - 28; http://dx.doi.org/10.1101/gad.1424806; PMID: 16751179
- Colella S, Shen L, Baggerly KA, Issa JP, Krahe R. Sensitive and quantitative universal Pyrosequencing methylation analysis of CpG sites. Biotechniques 2003; 35:146 - 50; PMID: 12866414
- Shen L, Kondo Y, Guo Y, Zhang J, Zhang L, Ahmed S, et al. Genome-wide profiling of DNA methylation reveals a class of normally methylated CpG island promoters. PLoS Genet 2007; 3:2023 - 36; http://dx.doi.org/10.1371/journal.pgen.0030181; PMID: 17967063
- Wang Z, Zang C, Cui K, Schones DE, Barski A, Peng W, et al. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell 2009; 138:1019 - 31; http://dx.doi.org/10.1016/j.cell.2009.06.049; PMID: 19698979