Abstract
The mechanisms by which the placenta adapts to exogenous stimuli to create a stable and healthy environment for the growing fetus are not well known. Low oxygen tension influences placental function, and is associated with preeclampsia, a condition displaying altered development of placental trophoblast. We hypothesized that oxygen tension affects villous trophoblast by modulation of gene expression through DNA methylation. We used the Infinium HumanMethylation450 BeadChip array to compare the DNA methylation profile of primary cultures of human cytotrophoblasts and syncytiotrophoblasts under < 1%, 8% and 20% oxygen levels. We found no effect of oxygen tension on average DNA methylation for either cell phenotype, but a set of loci became hypermethylated in cytotrophoblasts exposed for 24 h to < 1% oxygen, as compared with those exposed to 8% or 20% oxygen. Hypermethylation with low oxygen tension was independently confirmed by bisulfite-pyrosequencing in a subset of functionally relevant genes including CD59, CFB, GRAM3 and ZNF217. Intriguingly, 70 out of the 147 CpGs that became hypermethylated in < 1% oxygen overlapped with CpG sites that became hypomethylated upon differentiation of cytotrophoblasts into syncytiotrophoblasts. Furthermore, the preponderance of altered sites was located at AP-1 binding sites. We suggest that AP-1 expression is triggered by hypoxia and interacts with DNA methyltransferases (DNMTs) to target methylation at specific sites in the genome, thus causing suppression of the associated genes that are responsible for differentiation of villous cytotrophoblast to syncytiotrophoblast.
Keywords: :
Introduction
Epigenetic processes, including DNA methylation, play a role in programmed gene regulation during cellular differentiation and mediate adaptation to environmental conditions. Pregnancy is a condition where environmentally mediated epigenetic programming can influence long-term health and disease in both mother and offspring. The placenta plays a central role in modulating the environment to which the fetus is exposed, and many effects on the fetus may be the result of epigenetic changes to the placenta.Citation1,Citation2 In this study, we address epigenetic effects on the placenta by controlling oxygen tension as the independent variable, which has been implicated in the placental dysfunction typical of adverse pregnancy outcomes.
The placenta is pivotal for fetal growth and well-being during pregnancy. The trophoblast bi-layer regulates gene expression and metabolism in response to genetic and environmental signals, including levels of oxygen, nutrients and hormones. Oxygen tension plays a key role in placental development, where the pO2 is < 20 mmHg at the implantation site prior to 8–10 weeks but rises to 40–60 mmHg (5–8% oxygen) after 12 weeks gestation to continue at this level for the second and third trimesters.Citation3,Citation4 This rise in pO2 coincides with entry of maternal blood into the intervillous space between 10–12 weeks and perfusion of the tree-like chorionic villi that mediate maternal-fetal exchange. Villi are surfaced by a terminally differentiated syncytiotrophoblast, a true syncytium with multiple nuclei in the same cytoplasmic mass. A subjacent mononucleated cytotrophoblast layer is mitotically active and fuses to replenish the syncytiotrophoblast in a highly regulated process under the influence of oxygen tension. The villous core is bounded by the trophoblast basement membrane and consists of a mesenchymal connective tissue containing the fetal blood vessels. Underperfusion with hypoxia, re-perfusion with oxidative stress, or both associate with the placentas of women with preeclampsia (PE) and fetal intrauterine growth restriction (IUGR), yielding altered gene expression and placental pathology in these clinical maladies.Citation5
Epigenetic alterations play a central role in the cell’s response pathways that are critical for adaptation to hypoxia.Citation6 Hypoxia causes global hypomethylation in skin fibroblasts and in tumors (colorectal and melanoma cancer).Citation7 In contrast, hypoxia in prostate cells cultured > 24h yields a global increase in DNA methylation, including altered DNA methylation at many imprinted loci.Citation8 The role of epigenetic alterations upon hypoxia treatment in placenta has been implicated in the mouse.Citation9 Interestingly, hypomethylation of many gene promoters is observed in placentas associated with early-onset preeclampsia (EOPET).Citation10 We tested the hypothesis that oxygen levels modulate DNA methylation in cytotrophoblasts, syncytiotrophoblasts, or both phenotypes during primary culture of human trophoblasts. We further compared these to the DNA methylation differences arising as a consequence of differentiation from cytotrophoblast into syncytiotrophoblast in culture.
Results
Genome-wide DNA methylation analysis
Ninety trophoblast genomic DNA samples (30 test samples; each with 3 biological replicates), as illustrated in , were obtained and DNA methylation profiles were assessed using the Illumina Infinium Human Methylation 450 Bead Chip. To get an overall sense of the relationship of samples to each other we first used unsupervised hierarchical clustering of the samples based on their overall DNA methylation profiles in 453,244 probes (see Methods for quality control). Samples clustered by the placenta of origin rather than by culture conditions (< 1%, 8% and 20% O2) or cyto/syncytiotrophoblast differentiation (Fig. S1), suggesting that placenta-specific genetic and/or epigenetic differences (such as pregnancy-specific in utero effects on the epigenome) were the main source of variance among samples. The cluster pattern was unaltered by eliminating probes with known genetic polymorphisms at the target CpG. Using the array average as a measure of global methylation, we also found no difference in average methylation level among the exposure groups (Fig. S2). This shows that there are not global effects on the level of DNA methylation by treatment.
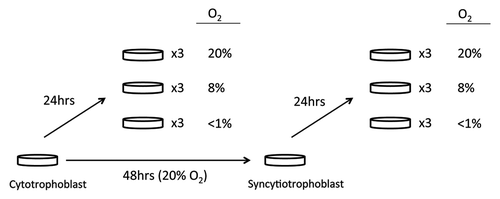
Figure 1. Cell culture condition for placental trophoblast samples. Cell cultures were plated and maintained in a culture support center at 37°C in a 5% carbon dioxide/air atmosphere (20% oxygen, standard conditions). After 4 h to allow attachment, half of the cells from each placenta were continued in standard conditions of 5% carbon dioxide/air for 48 h until exposure to < 1% oxygen conditions, 8% oxygen conditions or standard conditions, while the other half of the cells were exposed to < 1% oxygen, 8%, or standard conditions for 24 h.
DNA methylation changes with oxygen exposure
Given the tight clustering of samples by placenta of origin, we predicted that only a small proportion of loci would be altered due to oxygen tension or cell phenotype. To reduce the possibility of false positives, we used a relatively stringent cut-off of > 10% absolute average methylation difference (Δβ > 0.1 from Illumina array) and also required loci to be significant (p < 0.05) and show this magnitude of change (Δβ > 0.1) in each of three independent replicate experiments. We observed no significant difference in methylation for any CpG site by our criteria (p < 0.05 and Δβ > 0.1) when comparing 8% to 20% oxygen levels in either cytotrophoblast or syncytiotrophoblast. However, we found a cassette of loci with altered DNA methylation comparing low oxygen level (< 1%) to either 8% or 20% in both cell types. As many similar sites were significant in the 8% and the 20% vs. < 1% oxygen comparisons, we decided to reduce the potential for false positive findings by considering as “hypoxia-associated changes” only those methylation changes that were 1) significant when comparing 1% to both 8% and 20% oxygen and 2) shared in common between the three replicate experiments. Doing this, we identified 147 CpG loci among 83 genes that were consistently (in each of the three replicates) hypermethylated (average Δβ > 0.1; p < 0.05) in cytotrophoblasts after 24 h exposure to < 1% oxygen level (Fig. S3A and Table S1). In contrast, there were no differentially methylated loci related to oxygen exposure in syncytiotrophoblast cultures using the same criteria. Thus, the epigenetic response to hypoxia depends on trophoblast phenotype.
DNA methylation changes with cell phenotype
As hypoxia exposure can cause delayed differentiation of cytotrophoblasts into syncytiotrophoblast in culture, it was of interest to compare DNA methylation profile by cell phenotype to changes we observed with hypoxia exposure. Cellular differentiation is normally accompanied by epigenetic alterations, with a general tendency to acquire de novo DNA methylation marks. Considering only cells cultured at 8% oxygen (representing in vivo oxygen level for a term placenta), we found 223 loci that consistently showed a decreased methylation (average Δβ > 0.1) in syncytiotrophoblast, as compared with cytotrophoblasts, while no CpG site showed increased levels of methylation during this change in cellular phenotype (Fig. S3B andTable S2). Intriguingly, 70 out of 147 hypoxia-associated CpGs, which became hypermethylated in low oxygen, overlapped with CpG sites that became hypomethylated upon differentiation of cytotrophoblasts into syncytiotrophoblasts. When comparing the average methylation values for these overlapping sites, there was a trend toward less methylation in cells exposed to 20% as compared with 8% but with the lowest methylation values for syncytiotrophoblast (i.e., < 1% cytotrophoblast > 8% cytotrophoblast > 20% cytotrophoblast > 8% syncytiotrophoblast; Table S3). While the methylation at 20% oxygen was consistently lower than that for 8% oxygen, the differences were all < 10% explaining why these sites were not initially identified as significantly altered by our criteria requiring an average Δβ > 0.1 ( and ).
Figure 2A-B. DNA methylation patterns at each differentially methylated CpG and its nearby sites. Examples illustrated are (A) CD59 and (B) GRAMD3. Although only the CpG at the site of interest met the criteria for a statistically significant methylation difference (p < 0.05 and Δβ > 0.1), methylation differences are also observed at the nearby sites.
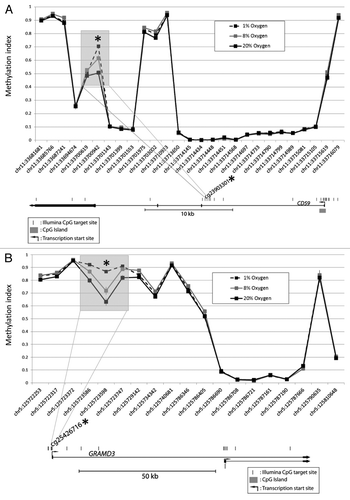
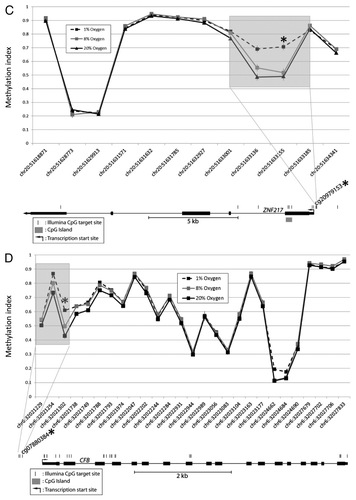
Figure 2C-D. DNA methylation patterns at each differentially methylated CpG and its nearby sites. Examples illustrated are (C) CFB and (D) ZNF217. Although only the CpG at the site of interest met the criteria for a statistically significant methylation difference (p < 0.05 and Δβ > 0.1), methylation differences are also observed at the nearby sites.
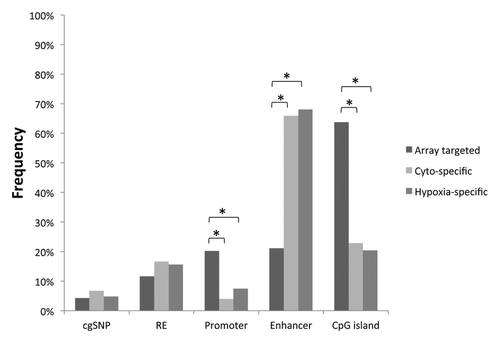
Possible confounders for results from array-based methylation technologies include the cross-hybridization of probes to multiple regions of the genome or the interference of hybridization due to probes being located on or near polymorphisms.Citation11,Citation12 However, only two probes in the differentially methylated regions identified as specific to cytotrophoblasts were likely to cross-hybridize into other regions of the genome (data not shown). We also found no enrichment of SNPs in the differentially methylated regions in either the phenotype-specific or the hypoxia-specific CpGs (). These findings excluded the possibility that the observed methylation changes were caused by DNA sequence bias in the probes. Moreover, DNA sequence bias was unlikely to play a role as the samples were genetically matched for all comparisons.
Figure 3. Genomic characteristics of differentially methylated regions. The distribution of CpG loci that were targeted by the methylation array (Array targeted), cytotrophoblast-specifically methylated (Cyto-specific) or hypoxia-specifically methylated (Hypoxia-specific) are shown with respect to different genomic characteristics. RE: repetitive element, *p < 0.0005.
Gene ontology analysis of DNA methylation changes
Ontology of the genes associated with the 147 hypoxia-associated CpGs in cytotrophoblasts showed enrichment for genes with functions involved in “signaling” and “phosphoproteins” (FDR < 10%; ). Possibly, phosphorylation based signaling networks are affected by oxygen tension. Ontology analysis of the genes with altered DNA methylation by cell-phenotype showed enrichment for genes that functioned in the plasma membrane (). This is consistent with the observation that hypoxia delays cytotrophoblast fusion,Citation13 and suggests that this delay involves silencing of genes via DNA methylation that otherwise would be expressed upon differentiation.
Table 1. Functions of differentially methylated loci between 8% and 20% vs. low (< 1%) oxygen levels
Table 2. Functions of genes associated in common between hypoxia- and cell-phenotype-differentially methylated loci
Genomic characteristics and motif analysis of DNA methylation changes
Sites of altered DNA methylation were investigated for their association with repetitive elements, promoters, enhancers and CpG islands. The probe distribution for these genomic features was very similar between phenotype-specific and hypoxia-specific differentially methylated regions (), which was consistent with the substantial overlap between the two groups. While there was no enrichment of repetitive elements, we found that the hypoxia associated probes were significantly less likely to be in promoter regions (p < 0.0005) and CpG islands (p < 0.0005), but they were significantly more likely to be in enhancer regions (p < 0.0005; ).
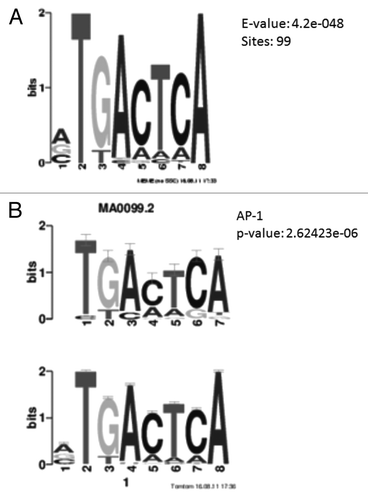
To gain a better knowledge of the mechanism involved in DNA methylation alteration upon hypoxia treatment, we performed a DNA motif search based on a 100 bp window on either side of the differentially methylated cytosine nucleotides. We found 99 of the 147 hypoxia-associated loci (~70%) were located within 100 bp of the consensus DNA sequence TGACTCA (enrichment or E-value = 4.2e-48). Similarly, the TGACTCA motif was also enriched in the cell-phenotype differentially methylated regions (42%, E-value = 4.3e-51). This sequence is the transcriptional binding motif of Activator Protein 1 (AP-1; ), a transcription factor composed of proteins from the c-Fos, c-Jun, ATF and JDP families.
Figure 4. Motif analysis of hypoxia differentially methylated loci. (A) Position weight matrix description of DNA sequence enriched in hypoxia differentially methylated loci. A consensus DNA sequence of TGACTCA is enriched in 99 out of the 147 loci. (B) The enriched DNA sequence matches the consensus sequence of AP-1 binding region.
Genome-wide expression analysis of cytotrophoblasts exposed to hypoxia
Although we assume a functional significance of AP-1 binding sites in hypoxia, the relationship between DNA methylation and gene expression of the sites remained to be determined. We thus performed a genome-wide expression study using quality checked mRNA extracted from the same cultures as used for DNA methylation studies. This included 15 cytotrophoblast samples with the three oxygen paradigms studied. We used a p < 0.05 and average absolute fold change of 20% to identify differentially expressed genes, given the small number of differentially methylated sites found and the relatively low absolute levels of DNA methylation. We identified 2034 genes differentially expressed among the samples < 1% vs. 8% and < 1% vs. 20% (data not shown).
Gene co-expression analysis showed that the 1,022 genes upregulated by hypoxia were also enriched in “phosphoproteins” (), while the 1,012 downregulated genes were enriched in “acetylation” (). However, only five of the 83 hypoxia-associated, hypermethylated genes showed the expected decreased expression using our criteria for altered expression (p < 0.05 and fold change of 20%). Collectively, these observations suggest that a change in DNA methylation is not generally the primary trigger of global gene expression changes observed with exposure of trophoblasts to hypoxia.
Table 3. Functions enriched for hypoxia-upregulated genes (redundant GO terms related to “ribosome,” “cytosol” and “cell death” were excluded)
Table 4. Functions enriched for hypoxia-downregulated genes (redundant GO terms related to “organelle lumen,” “mitochondrion” and “RNA-binding” were excluded)
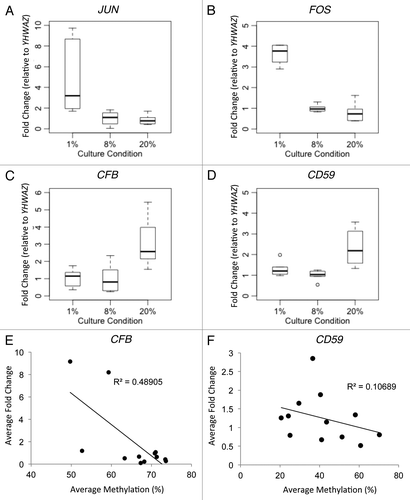
Since AP-1 binding sites were enriched in the vicinity of differentially methylated CpGs in response to hypoxia, the gain of DNA methylation may be mediated by increased expression of AP-1 genes. Indeed, both JUN and FOS showed significantly higher expression in the < 1% oxygen group compared with the other two oxygen levels (JUN p < 0.0005, FOS p < 0.005) ( and B), while HIF1A expression did not increase significantly.
Figure 5. Gene expression and DNA methylation correlation of candidate genes. Gene expression as validated by Realtime RT-PCR was shown for (A) JUN, (B) FOS, (C) CFB and (D) CD59. Since the promoters of JUN and FOS are unmethylated, DNA methylation correlation with gene expression was shown only for (E) CFB and (F) CD59.
Confirmation of array results by bisulfite pyrosequencing and reverse-transcription real-time PCR
We selected six genes that were functionally relevant to placenta from the array data for confirmatory studies using targeted approaches. Using bisulfite pyrosequencing, we observed no DNA methylation at the promoter regions of JUN and FOS in any trophoblast sample (Fig. S4Aand B), while real-time reverse-transcription PCR confirmed the increased gene expression of JUN and FOS at < 1% vs. 8% and 20% oxygen ( and B). Thus, expression levels of these genes do not appear to be regulated by promoter DNA methylation in cultured trophoblasts. Pyrosequencing confirmed that the gene promoters GRAMD3, CFB, CD59 and ZNF217 showed increased methylation in < 1% vs. 8% oxygen (p < 0.01 for all four loci) and significantly lower methylation in 20% vs. 8% oxygen (p < 0.01 for all except p < 0.05 for CFB) (Fig. S4C-F). The expression of CFB and CD59 were decreased in < 1% and 8% vs. 20% oxygen ( and D) and their expression levels were inversely correlated with DNA methylation levels ( and F). Thus DNA methylation is associated with the expression levels of only a subset of genes with altered expression, as expected.
Discussion
This is the first genome-wide analysis of the effect of phenotype and oxygen concentration on DNA methylation and gene expression of human villous cytotrophoblasts and syncytiotrophoblasts. The data show that cytotrophoblasts differ from syncytiotrophoblasts in their methylation profile under standard culture conditions and in response to hypoxia. There are substantial changes of DNA methylation at enhancers of genes responsible for signaling in cytotrophoblast upon hypoxic exposure. However, these responses are unlikely to be the primary mechanism for the majority of gene expression changes observed. Instead, the methylation changes can be attributed to upregulation of genes coding for proteins that comprise the transcription factor AP-1. We speculate that binding of AP-1 to specific sequences can increase DNA methylation and inhibit transcription at these regions and that the methylation of gene promoters or enhancers may play a role in impeding differentiation of villous trophoblasts.
Hypoxia causes global hypomethylation in skin fibroblasts, colorectal tumors and melanomas.Citation7 In contrast, hypoxia in cultured prostate cells yields global increases in DNA methylation and altered DNA methylation at imprinted loci, possibly as a result of increased expression of DNMT3b.Citation8 We observed a much more limited effect on DNA methylation in human trophoblast cultures and specifically, there was no significant change in global DNA methylation as assessed by overall average DNA methylation in the Illumina array. Similarly, there was a relatively small set of loci that showed loss of DNA methylation in the process of differentiation from cytotrophoblast to syncytiotrophoblast. The evidence for DNA demethylases is controversial and loss of DNA methylation is normally thought to occur by passive, replication-dependent manner (i.e., failure to methylate hemi-methylated DNA after replication).Citation14 However, differentiation of cytotrophoblast to syncytiotrophoblast occurs through cell fusion, not cell division, and hence, the altered DNA methylation must have occurred through a replication-independent mechanism.
Interestingly, all loci with significantly altered DNA methylation at low levels of oxygen showed an increase of DNA methylation, while no locus showed significant decreases in methylation. The effects were greatest between < 1% and 20% oxygen, but there was also a trend for less methylation in 8% as compared with 1% oxygen at these same loci. The hypoxia changes observed in cytotrophoblasts included regions associated with several genes relevant to the placenta. For example, CP, coding for ceruloplasmin, and ITGA5, coding for α-5 integrin, are reported to show increased expression in placentas from preeclampsia and in trophoblast cultures grown in low oxygen.Citation15,Citation16 Other genes in the vicinity of the CpGs with altered methylation are reported to show altered gene expression in response to hypoxia, including SOD2,Citation17 XDHCitation18,Citation19 and ZNF217.Citation20 Collectively, these hypoxia-induced, differentially methylated genes are enriched for signaling function (), which is not a feature shared with cell-phenotype-differentially methylated loci. This suggests that genes associated with differentially methylated loci in response to hypoxia may have secondary effects on genome-wide expression.
We observed an extensive change of genome-wide gene expression in cytotrophoblasts exposed to hypoxia (using a 20% change cut-off), which is consistent with previous studies and more prominent than that observed for DNA methylation. Notably, many of the upregulated genes were associated with cell death, a response known to occur in trophoblasts exposed to very low oxygen tensions.Citation21 Among the genes upregulated > 4-fold in < 1% vs. 8% oxygen were VEGFA and the transcription factors JUN and FOS. Moreover, genes exhibiting decreased expression (< 25%) in response to < 1% vs. 8% oxygen were genes from a number of histone proteins, such as HIST1H2BG, HIST1H3D, HIST1H4 and HIST2H2BE. This may explain why gene ontogeny analysis revealed “acetylation” as being prominent in the hypoxia-downregulated genes.
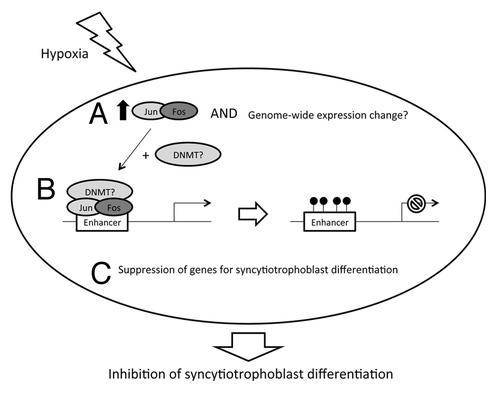
Interestingly, many regions that showed increased methylation after hypoxia exposure corresponded to regions that showed decreased methylation upon differentiation into syncytiotrophoblast. As cytotrophoblast cultured in hypoxic conditions show delayed fusion to syncytiotrophoblast,Citation13 we speculate that this is in part due to active suppression of genes by DNA methylation that would otherwise be upregulated upon differentiation. Intriguingly, such inhibition of villous trophoblast differentiation has been observed in placentas from preeclamptic women where malperfusion is common.Citation22 Chronic hypoxia in prostate cells was associated with increased expression of the DNA-methyltransferase, DNMT3B, which was argued to explain gene-specific changes in DNA methylation.Citation8 Nonetheless, we suggest that the observed methylation changes in trophoblast cells may occur as a consequence of chromatin alterations occurring as a consequence of altered gene expression, such as changes to chromatin resulting from binding of proteins to DNA. The hypoxia-associated loci tend to be located in enhancer rather than promoter or CpG island regions. Through motif analysis, we found that 70% of the hypoxia-associated loci were located in the transcriptional binding motif of AP-1, which is composed of transcriptional binding proteins such as c-Fos and c-Jun (). AP-1 is known to regulate gene expression in response to a variety of stimuli, including cytokines, growth factors, stress and bacterial and viral infections, which in turn control a number of cellular processes including differentiation, proliferation and apoptosis. AP1-has previously been reported to downregulate genes through recruitment of histone deacetylases.Citation23,Citation24 We propose that expression of AP-1 is triggered by hypoxia, and this may either interact with DNA methyltransferases (DNMTs) to target methylation at specific sites in the genome, or DNA methylation occurs subsequent to histone deactylation; which then causes suppression of the associated genes that are responsible for syncytiotrophoblast differentiation (). Further molecular studies, such as chromatin immunoprecipitation sequencing, to confirm AP-1 and enhancer sequence interaction for the associated genes are needed to confirm this model.
Figure 6. Proposed model of hypoxia effect on cytotrophoblast. Hypoxic exposure may trigger (A) increased expression of JUN and FOS (and perhaps a genome-wide change of gene expression) which encodes Jun and fos to form AP-1 proteins. (B) AP-1 may then recruit DNA methylation machinery (such as DNMTs) for de novo methylation of CpGs at the enhancer regions of various genes. (C) This may cause a suppression of expression for genes that are responsible for syncytiotrophoblast differentiation, which results in depletion of syncytiotrophoblast formation.
Methods
Isolation and culture of primary human trophoblasts
This study was approved by the Institutional Review Board of Washington University School of Medicine in St. Louis, MO and University of British Columbia, Vancouver, BC. Primary human trophoblasts (n = 5) were isolated from uncomplicated singleton pregnancies delivered by repeat cesarean section at 39–40 weeks’ gestation using the trypsin-deoxyribo-nuclease-dispase/Percoll method, as previously described.Citation13,Citation21,Citation25 All experiments were performed in triplicate (i.e., each placenta was split into three samples for each treatment). There was a very high degree of overlap of hypoxia associated changes and replicate experiments were used to reduce risk of false-positive results. Cultures were plated at a density of 300,000 cells/cm2 and maintained in Dulbecco's modified Eagle's medium (Sigma, St. Louis, MO) containing 10% fetal bovine serum (Gibco, Grand Island, NY), 20 mmol/liter HEPES pH 7.4 (Sigma), penicillin (100 units/ml), streptomycin (100 μg/ml) and fungizone (0.25 μg/ml; all from Washington University Tissue Culture Support Center) at 37°C in a 5% carbon dioxide/air atmosphere (20% oxygen, standard culture conditions). After 4 h to allow attachment, as illustrated in , half of the cells (cytotrophoblasts) were exposed to < 1% oxygen (< 1% O2/5%CO2/10%H2/balance N2), 8% oxygen (8% O2/5%CO2/10%H2/balance N2) or standard culture conditions, for 24 h. The other half of the cells from each placenta were continued in standard conditions for 48 h, confirmed the successful formation of syncytiotrophoblasts (> 85% of cells are multinucleatedCitation26) using immunofluorescence staining for E-cadherin, which stains plasma membranes among trophoblasts (data not shownCitation26-Citation28). These syncytiotrophoblasts were then exposed to < 1% oxygen, 8% oxygen or standard culture conditions (5%CO2 in air with 20% oxygen) for 24 h.
Genomic DNA and total RNA extraction
Genomic DNA was purified from cultured trophoblasts using Wizard® Genomic DNA Purification Kit (Promega, Madison, WI), and Total RNA was purified using TriReagent (Molecular Research Center, Inc., Cincinnati, OH), according to the manufacturer's instructions.
Illumina HumanMethylation450 array
The Illumina HumanMethylation450 array quantifies methylation at 485,512 CpGs in > 20,000 genes. All samples were run with three replicates on the array in the same batch to avoid any potential bias due to batch effect. Raw data was first subject to (1) background subtraction; (2) elimination of probes with detection p value > 0.01 (453,233 probes remained for analysis); (3) measurements were converted to an “M-value” (log2 ratio of the intensities of methylated to unmethylated probes)Citation29; (4) Color channel bias adjustment; and (5) Quantile normalization. Candidate sites were prioritized by (1) p-value < 0.05 (Student’s T-test) in each of three replicate experiments; (2) Magnitude of difference between group means >10% (i.e. Δβ > 0.1), which is more likely to be biological meaningfulCitation30; (3) the detection of multiple altered CpGs associated with the same gene; and (4) potential role in trophoblast function and/or hypoxia response.
Bisulfite pyrosequencing
DNA methylation of selected hypoxia differentially methylated loci was confirmed using bisulfite pyrosequencing. DNA was bisulfite converted and pyrosequencing was performed on a Biotage Pyromark Q96 MD Pyrosequencer. The quantitative levels of methylation for the CpG dinucleotide were evaluated with the Pyro Q-CpG software (Biotage, Uppsala, Sweden). Pyrosequencing assays were designed for FOS, JUN, CD59, CFB, GRAM3 and ZNF217 (Table S4). Methylation-unbiased pyrosequencing primers were designed to include the same CpG sites that were interrogated by the Illumina probes, and each assay resulted in methylation levels that were highly correlated with those estimated by the Illumina array (R > 0.9).
Illumina HumanHT-12 v4 expression array
RNA extracted from cytotrophoblast exposed to different oxygen conditions was used for the expression array study. Fifteen cDNA samples (5 in < 1%, 6 in 8% and 4 in 20% oxygen exposure group from 2 different placental samples) that passed quality control were hybridized to the Illumina HumanHT-12 v4 Expression BeadChip (Illumina, San Diego, CA), which assessed gene expression of > 24,000 genes using 47,231 probes. Raw data was processed with background subtraction and quantile normalization. Probes with a detection p value > 0.01 were eliminated.
Real time RT-PCR
The gene expression of CD59 (Hs00174141_m1), JUN (Hs99999141_s1), FOS (Hs01119267_g1) and CFB (Hs01006494_g1) was validated using targeted Taqman Gene Expression assays (Applied Biosystems, Melbourne, Australia). YWHAZ (Hs03044281_g1) was used as an endogenous control, as it is known to be highly expressed in placenta while not differing with gestational age.Citation31 For each sample, 500 ng of RNA was reverse transcribed using the QuantiTect Reverse Transcription Kit (Qiagen, Hilden, Germany). Each assay was run with cDNA samples, non-template control and calibrator in triplicate using an ABI7500 Real time Thermal Cycler (Applied Biosystems, Melbourne, Australia). A total PCR reaction volume of 20 ul in each well was run with a 1:10 cDNA dilution. Relative quantification was calculated using the ∆∆CT method. Briefly, the difference between CT values (ΔCT) for the target genes and endogenous control was compared with the calibrator for each plate. The calibrator was composed of pooled cDNA from the 8% oxygen exposure group. While FOS, JUN and CFB have a good correlation of gene expression between values from qPCR and array (R > 0.7), the correlation between the two methods was low for CD59 (R = 0.33), which may due to the milder change of expression compared with other genes. In addition to the 15 samples run on expression array, 21 more RNA samples from cytotrophoblasts of 2 other placentas run in the methylation array were assessed for gene expression of the 4 genes.
Bioinformatic analysis
The Database for Annotation, Visualization and Integrated Discovery (DAVID) (http://david.abcc.ncifcrf.gov/) was used for gene ontology (GO) analysis. Functions of genes with differential methylation or expression were compared against those represented on the array. Enriched GO terms were identified using a cut-off of FDR < 10%. The Multiple Em for Motif Elicitation (MEME) site (http://meme.sdsc.edu/) was used to identify motifs enrichment on DNA sequences. Motif search was performed on a 100 bp window with the CpG site of interest at center. Enriched sequences were then compared with all known motifs in a database.
Statistical analysis of genomic features
Cross-hybridization of probes was investigated by the BLAT function in UCSC browser. Information on SNPs, repetitive elements, promoter, enhancer and CpG island for each probe was extracted from Illumina annotation. A Chi-square test was performed to determine the enrichment or reduction for candidate loci on different genomic features.
Additional material
Download Zip (227.4 KB)Acknowledgments
We would like to thank Dr Maria S. Peñaherrera, Ruby Jiang and Magda Price for technical support. We would like to thank Dr Mike Kobor, Lucia Lam and Sarah Neumann for use of equipment involved in microarray processing and for use of the Biotage PyroMarkTM MD machine. This work was supported by the Canadian Institutes for Health Research (Funding Reference Number 119402, W.P.R.); The Barnes-Jewish Hospital Foundation, D.M.N.; and NIH HD 29190, D.M.N.)
Author Contributions
R.K.C.Y., W.P.R. and D.M.N. conceived the study and wrote the draft manuscript. R.K.C.Y., B.C. and J.B. designed and performed the experiments. R.K.C.Y., B.C. and J.B. analyzed the data. DMN contributed the tissue samples. All authors participated in critical discussion, wrote the manuscript and approved the final manuscript.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Hogg K, Price EM, Hanna CW, Robinson WP. Prenatal and perinatal environmental influences on the human fetal and placental epigenome. Clin Pharmacol Ther 2012; 92:716 - 26; http://dx.doi.org/10.1038/clpt.2012.141; PMID: 23047650
- Novakovic B, Saffery R. The ever growing complexity of placental epigenetics - Role in adverse pregnancy outcomes and fetal programming. Placenta 2012; 33:959 - 70; http://dx.doi.org/10.1016/j.placenta.2012.10.003; PMID: 23102655
- Schneider H. Oxygenation of the placental-fetal unit in humans. Respir Physiol Neurobiol 2011; 178:51 - 8; http://dx.doi.org/10.1016/j.resp.2011.05.009; PMID: 21621012
- Tuuli MG, Longtine MS, Nelson DM. Review: Oxygen and trophoblast biology--a source of controversy. Placenta 2011; 32:Suppl 2 S109 - 18; http://dx.doi.org/10.1016/j.placenta.2010.12.013; PMID: 21216006
- Kingdom J, Huppertz B, Seaward G, Kaufmann P. Development of the placental villous tree and its consequences for fetal growth. Eur J Obstet Gynecol Reprod Biol 2000; 92:35 - 43; http://dx.doi.org/10.1016/S0301-2115(00)00423-1; PMID: 10986432
- Watson JA, Watson CJ, McCann A, Baugh J. Epigenetics, the epicenter of the hypoxic response. Epigenetics 2010; 5:293 - 6; http://dx.doi.org/10.4161/epi.5.4.11684; PMID: 20418669
- Shahrzad S, Bertrand K, Minhas K, Coomber BL. Induction of DNA hypomethylation by tumor hypoxia. Epigenetics 2007; 2:119 - 25; http://dx.doi.org/10.4161/epi.2.2.4613; PMID: 17965619
- Watson JA, Watson CJ, McCrohan AM, Woodfine K, Tosetto M, McDaid J, et al. Generation of an epigenetic signature by chronic hypoxia in prostate cells. Hum Mol Genet 2009; 18:3594 - 604; http://dx.doi.org/10.1093/hmg/ddp307; PMID: 19584087
- Gheorghe CP, Mohan S, Oberg KC, Longo LD. Gene expression patterns in the hypoxic murine placenta: a role in epigenesis?. Reprod Sci 2007; 14:223 - 33; http://dx.doi.org/10.1177/1933719107302860; PMID: 17636235
- Yuen RK, Peñaherrera MS, von Dadelszen P, McFadden DE, Robinson WP. DNA methylation profiling of human placentas reveals promoter hypomethylation of multiple genes in early-onset preeclampsia. Eur J Hum Genet 2010; 18:1006 - 12; http://dx.doi.org/10.1038/ejhg.2010.63; PMID: 20442742
- Blair JD, Price EM. Illuminating potential technical artifacts of DNA-methylation array probes. Am J Hum Genet 2012; 91:760 - 2; http://dx.doi.org/10.1016/j.ajhg.2012.05.028; PMID: 23040498
- Chen YA, Choufani S, Grafodatskaya D, Butcher DT, Ferreira JC, Weksberg R. Cross-reactive DNA microarray probes lead to false discovery of autosomal sex-associated DNA methylation. Am J Hum Genet 2012; 91:762 - 4; http://dx.doi.org/10.1016/j.ajhg.2012.06.020; PMID: 23040499
- Nelson DM, Johnson RD, Smith SD, Anteby EY, Sadovsky Y. Hypoxia limits differentiation and up-regulates expression and activity of prostaglandin H synthase 2 in cultured trophoblast from term human placenta. Am J Obstet Gynecol 1999; 180:896 - 902; http://dx.doi.org/10.1016/S0002-9378(99)70661-7; PMID: 10203658
- Cedar H, Bergman Y. Programming of DNA methylation patterns. Annu Rev Biochem 2012; 81:97 - 117; http://dx.doi.org/10.1146/annurev-biochem-052610-091920; PMID: 22404632
- Zhao H, Jiang Y, Cao Q, Hou Y, Wang C. Role of integrin switch and transforming growth factor Beta 3 in hypoxia-induced invasion inhibition of human extravillous trophoblast cells. Biol Reprod 2012; 87:47; http://dx.doi.org/10.1095/biolreprod.112.099937; PMID: 22674391
- Guller S, Buhimschi CS, Ma YY, Huang ST, Yang L, Kuczynski E, et al. Placental expression of ceruloplasmin in pregnancies complicated by severe preeclampsia. Lab Invest 2008; 88:1057 - 67; http://dx.doi.org/10.1038/labinvest.2008.74; PMID: 18679377
- Nanduri J, Makarenko V, Reddy VD, Yuan G, Pawar A, Wang N, et al. Epigenetic regulation of hypoxic sensing disrupts cardiorespiratory homeostasis. Proc Natl Acad Sci U S A 2012; 109:2515 - 20; http://dx.doi.org/10.1073/pnas.1120600109; PMID: 22232674
- Budhiraja R, Kayyali US, Karamsetty M, Fogel M, Hill NS, Chalkley R, et al. Estrogen modulates xanthine dehydrogenase/xanthine oxidase activity by a receptor-independent mechanism. Antioxid Redox Signal 2003; 5:705 - 11; http://dx.doi.org/10.1089/152308603770380007; PMID: 14588143
- Wang G, Qian P, Jackson FR, Qian G, Wu G. Sequential activation of JAKs, STATs and xanthine dehydrogenase/oxidase by hypoxia in lung microvascular endothelial cells. Int J Biochem Cell Biol 2008; 40:461 - 70; http://dx.doi.org/10.1016/j.biocel.2007.08.008; PMID: 17920330
- Mao XG, Yan M, Xue XY, Zhang X, Ren HG, Guo G, et al. Overexpression of ZNF217 in glioblastoma contributes to the maintenance of glioma stem cells regulated by hypoxia-inducible factors. Lab Invest 2011; 91:1068 - 78; http://dx.doi.org/10.1038/labinvest.2011.56; PMID: 21483406
- Levy R, Smith SD, Chandler K, Sadovsky Y, Nelson DM. Apoptosis in human cultured trophoblasts is enhanced by hypoxia and diminished by epidermal growth factor. Am J Physiol Cell Physiol 2000; 278:C982 - 8; PMID: 10794672
- Redman CW, Sargent IL. Latest advances in understanding preeclampsia. Science 2005; 308:1592 - 4; http://dx.doi.org/10.1126/science.1111726; PMID: 15947178
- Kim LK, Choi UY, Cho HS, Lee JS, Lee WB, Kim J, et al. Down-regulation of NF-kappaB target genes by the AP-1 and STAT complex during the innate immune response in Drosophila. PLoS Biol 2007; 5:e238; http://dx.doi.org/10.1371/journal.pbio.0050238; PMID: 17803358
- Mittelstadt ML, Patel RC. AP-1 mediated transcriptional repression of matrix metalloproteinase-9 by recruitment of histone deacetylase 1 in response to interferon β. PLoS One 2012; 7:e42152; http://dx.doi.org/10.1371/journal.pone.0042152; PMID: 22879913
- Roh CR, Budhraja V, Kim HS, Nelson DM, Sadovsky Y. Microarray-based identification of differentially expressed genes in hypoxic term human trophoblasts and in placental villi of pregnancies with growth restricted fetuses. Placenta 2005; 26:319 - 28; http://dx.doi.org/10.1016/j.placenta.2004.06.013; PMID: 15823618
- Chen B, Longtine MS, Sadovsky Y, Nelson DM. Hypoxia downregulates p53 but induces apoptosis and enhances expression of BAD in cultures of human syncytiotrophoblasts. Am J Physiol Cell Physiol 2010; 299:C968 - 76; http://dx.doi.org/10.1152/ajpcell.00154.2010; PMID: 20810912
- Longtine MS, Chen B, Odibo AO, Zhong Y, Nelson DM. Caspase-mediated apoptosis of trophoblasts in term human placental villi is restricted to cytotrophoblasts and absent from the multinucleated syncytiotrophoblast. Reproduction 2012; 143:107 - 21; http://dx.doi.org/10.1530/REP-11-0340; PMID: 22046053
- Longtine MS, Chen B, Odibo AO, Zhong Y, Nelson DM. Villous trophoblast apoptosis is elevated and restricted to cytotrophoblasts in pregnancies complicated by preeclampsia, IUGR, or preeclampsia with IUGR. Placenta 2012; 33:352 - 9; http://dx.doi.org/10.1016/j.placenta.2012.01.017; PMID: 22341340
- Du P, Zhang X, Huang CC, Jafari N, Kibbe WA, Hou L, et al. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics 2010; 11:587; http://dx.doi.org/10.1186/1471-2105-11-587; PMID: 21118553
- Bibikova M, Lin Z, Zhou L, Chudin E, Garcia EW, Wu B, et al. High-throughput DNA methylation profiling using universal bead arrays. Genome Res 2006; 16:383 - 93; http://dx.doi.org/10.1101/gr.4410706; PMID: 16449502
- Murthi P, Fitzpatrick E, Borg AJ, Donath S, Brennecke SP, Kalionis B. GAPDH, 18S rRNA and YWHAZ are suitable endogenous reference genes for relative gene expression studies in placental tissues from human idiopathic fetal growth restriction. Placenta 2008; 29:798 - 801; http://dx.doi.org/10.1016/j.placenta.2008.06.007; PMID: 18684503