Abstract
Intrauterine nutrition can program metabolism, creating stable changes in physiology that may have significant health consequences. The mechanism underlying these changes is widely assumed to involve epigenetic changes to the expression of metabolic genes, but evidence supporting this idea is limited. Here we have performed the first study of the epigenomic consequences of exposure to maternal obesity and diabetes. We used a mouse model of natural-onset obesity that allows comparison of genetically identical mice whose mothers were either obese and diabetic or lean with a normal metabolism. We find that the offspring of obese mothers have a latent metabolic phenotype that is unmasked by exposure to a Western-style diet, resulting in glucose intolerance, insulin resistance and hepatic steatosis. The offspring show changes in hepatic gene expression and widespread but subtle alterations in cytosine methylation. Contrary to expectation, these molecular changes do not point to metabolic pathways but instead reside in broadly developmental ontologies. We propose that, rather than being adaptive, these changes may simply produce an inappropriate response to suboptimal environments; maladaptive phenotypes may be avoidable if postnatal nutrition is carefully controlled.
Introduction
Obesity and its accompanying morbidities, such as type 2 diabetes, are now pandemic. While there is no doubt that obesity has a genetic component, its rapidly increasing prevalence indicates a major environmental contribution; the etiology and familial patterns of obesity and diabetes are thus of great interest. Extensive evidence indicates that maternal nutrition and metabolism can stably affect the phenotypes of offspring, a phenomenon termed fetal programming.Citation1,Citation2 Rodent models of maternal overnutrition—diet-induced obesity and diabetes (through high fat and/or sugar diets), genetic manipulation (e.g., ob or db mice) and chemical/surgical induction of diabetesCitation1,Citation3,Citation4—show a variety of metabolic consequences in offspring, including increased body weight, adiposity, hypertension, insulin resistance, hyperglycemia, hyperphagia and leptin resistance (reviewed in refs. Citation1 and Citation3). Of these models, diet-induced maternal obesity and diabetes is most likely to reflect the metabolic syndrome seen in human populations but cannot distinguish the effect of excess fat consumption from the effect of maternal obesity and diabetes per se. Increasing the fat content of a diet also necessitates a relative reduction of other components in the diet. GeneticCitation5 and pairwise high fat-feeding modelsCitation6 have suggested that maternal obesity per se may cause programming effects that are distinct from those of dietary fat intake.
Since fetal programming appears to involve stable changes in phenotype, it has been widely speculated that epigenetic modifications induced by the intrauterine environment are responsible.Citation1,Citation2,Citation7 Epigenetic modifications specify stable states of gene expression; they are laid down largely during early development and remain relatively fixed over a lifetime (and sometimes into succeeding generations). In this view, fetal programming stems from induced epigenetic changes in genes that regulate metabolic pathways and has an adaptive function. But evidence for such targeted epigenetic change is scarce, and studies coupling the physiological and epigenomic consequences of exposure to fetal overnutrition, particularly in models of natural-onset obesity and diabetes, are lacking. It is at present unclear to what extent maternal obesity impacts on offspring phenotype via changes to epigenetic states.
We have previously characterized changes in cytosine methylation patterns in isogenic C57BL/6J mice in response to dietary intervention.Citation8 Cytosine methylation is a key epigenetic modification in vertebrates, acting as a focal point for mechanisms that suppress gene expression.Citation9 We found spontaneous and widespread methylation variability in genes with functions related to gene expression and development; similar variation has also been observed in humansCitation10 and methylation at four such methylation-variable loci appear to correlate with body mass index.Citation11 We found that exposure to dietary methyl donors dramatically increases the extent of this epigenetic variability.Citation8 We therefore wondered whether similar epigenomic effects would be manifest with exposure to maternal obesity.
Here we have studied the phenotypic and epigenomic effects of maternal obesity in a model of natural-onset obesity and type 2 diabetes, the Avy mouse. Our system allows comparison of genetically identical mice whose mothers either did, or did not, acquire diabetes and obesity eating a standard chow diet. We find that the offspring of obese mothers have a cryptic phenotype that is unmasked by exposure to a Western-style high fat diet: they remain nearly normal on a standard diet, but on a high fat diet they develop insulin resistance, glucose intolerance and hepatic steatosis. This predisposition is associated with gene expression and epigenomic changes affecting a broad variety of functions.
Results
A model of natural-onset maternal obesity
To observe the phenotypic and epigenomic consequences of natural-onset maternal obesity and diabetes, we exploited the obese yellow phenotype exhibited by Avy mice.Citation12,Citation13 The Avy allele is unusual in that it is subject to stochastic epigenetic silencing in an isogenic background, so that genetically identical mice can display markedly different phenotypes. Mice carrying an active allele have a characteristic syndrome of fully yellow pelage, obesity and type 2 diabetes; mice carrying a silent allele have agouti fur and a metabolically normal phenotype, termed pseudoagouti.Citation14 The Avy allele is congenic with nonagouti (a), and Avy mice are typically maintained as Avy/a heterozygotes; in a cross of an Avy/a dam with an a/a sire, 50% of offspring will be heterozygous for the Avy allele, and the other 50% will be homozygous a/a, on an isogenic C57BL/6J background.
In this experiment, we compared the a/a offspring of obese yellow diabetic Avy/a dams with genetically identical a/a offspring of lean pseudoagouti Avy/a dams (). In this way, phenotypic and epigenomic parameters could be assessed without confounding genetic variation. Furthermore, the induction of obesity and diabetes in yellow Avy mice does not involve any external manipulation: the mice are hyperphagic and by 3 mo of age they are obese, demonstrably hyperglycemic, hyperinsulinemic and insulin resistant; thus yellow coat color is inseparable from type 2 diabetes in adult Avy mice.Citation13,Citation15-Citation17 We chose the Avy model because the progressive development of metabolic syndrome in yellow Avy mice more accurately recapitulates the gradual onset of metabolic disease in humans with adult-onset obesity and diabetes. This is in contrast to genetic models of obesity and diabetes, which produce early-onset metabolic disease,Citation16,Citation18 or dietary manipulation where animals are forced onto a hypercaloric diet.
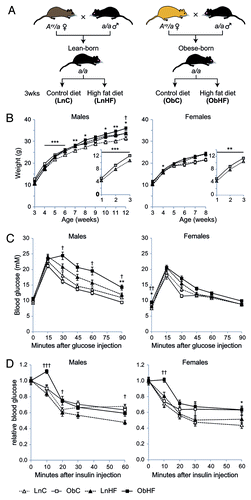
Figure 1. Offspring exposed to maternal obesity and diabetes have a latent predisposition to metabolic disease, which is revealed by a high-fat Western-style diet. (A) Experimental strategy. (B) Body weights of male and female offspring in each of the four groups in (A) (male: LnC n = 14, ObC n = 13, LnHF n = 15, ObHF n = 9; female: LnC n = 19, ObC n = 13, LnHF n = 13, ObHF n = 18); inset, pre-weaning weights for Lean-born and Obese-born animals. (C) Blood glucose levels in male and female animals during a glucose tolerance test at 6 weeks of age (male: LnC n = 14, ObC n = 13, LnHF n = 15, ObHF n = 9; female: LnC n = 20, ObC n = 6, LnHF n = 13, ObHF n = 11). (D) Blood glucose levels in animals during an insulin tolerance test at 7 weeks of age (male: LnC n = 14, ObC n = 17, LnHF n = 19, ObHF n = 19; female: LnC n = 20, ObC n = 13, LnHF n = 13, ObHF n = 18). Data are represented as mean ± SEM. Statistically significant differences are as indicated: *p < 0.05, **p < 0.01, ***p < 0.001 (LnC vs ObC); †p < 0.05, ††p < 0.01, †††p < 0.001 (LnHF vs ObHF).
A Western-style diet precipitates metabolic disease in obese-born offspring
In contrast to the overt defects in glucose metabolism seen in offspring of rodents fed a hypercaloric diet,Citation19-Citation21 mice born of obese yellow Avy/a mothers showed no evidence of disturbed glucose homeostasis when compared with offspring of lean Avy/a mothers (; Fig. S1). However, offspring of obese mothers (ObC) were slightly but significantly heavier than offspring of lean mothers (LnC) before weaning, and males remained so afterwards (). We asked if this indicated a cryptic or latent predisposition to metabolic disease that might be unmasked by a dietary challenge. We exposed both groups to a Western-style high fat diet and found that while both groups (LnHF and ObHF) gained more weight on this diet, male ObHF mice became significantly heavier than LnHF by 12 weeks (). After only three weeks on the Western diet (at 6 weeks of age) male ObHF mice demonstrated glucose intolerance () and insulin resistance (); their female siblings displayed slight insulin resistance but no impaired response to glucose (). Male (and to a lesser extent female) ObHF mice also exhibited a spike in blood glucose soon after intraperitoneal injection with insulin, indicating an impairment in their insulin response ().
To further assess the metabolic phenotype induced by natural-onset maternal diabetes and obesity, we sacrificed animals at 12 weeks of age for tissue analysis. We analyzed only male offspring as they demonstrated the most marked impairment in metabolic homeostasis. Liver lipidomic profiling revealed that total triacylglycerides (TAGs) and diacylglycerides (DAGs) were significantly higher in offspring of obese mothers, and levels were further elevated by the Western diet (; Fig. S2). H&E stained sections of liver were scored using pathological criteria for nonalcoholic steatohepatitis in humans.Citation22 The Western diet induced steatosis, which was markedly more severe in ObHF mice ().
Figure 2. Offspring exposed to maternal obesity and diabetes have defects in lipid metabolism which are exacerbated by a Western diet. Total liver (A) triacylglycerol and (B) diacylglycerol levels (LnC n = 4, ObC n = 6, LnHF n = 12, ObHF n = 8), and (C) steatosis scores (LnC n = 4, ObC n = 9, LnHF n = 10, ObHF n = 10) in livers of 12 week old male animals. Representative liver histology sections are shown in (D) (scale bar, 100 μm). (E) Serum leptin levels and (F) relationship between serum leptin levels and body weight at 12 weeks (LnC n = 6, ObC n = 7, LnHF n = 12, ObHF n = 12). Box and whisker plots show the median, 25th and 75th percentile values with whiskers indicating the maximum and minimum; means are indicated by an X. Statistically significant differences are indicated by asterisks: *p < 0.05, **p < 0.01, ***p < 0.001. Right-tailed Fisher’s exact test, LnHF c.f. ObHF steatosis scores: p = 0.012.
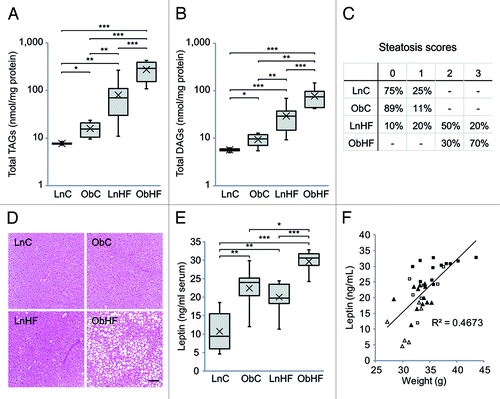
We also assessed serum leptin, an adipokine that circulates at levels proportional to body fat.Citation23 Leptin was higher in ObC mice than controls () and was further elevated by the Western diet. It was also raised by the Western diet in offspring of lean mothers. Leptin correlated with body mass in all of the groups (), suggesting that the observed increases in body weight are due to an increase in adiposity rather than an increase in lean mass.
Taken together, these results indicate that the offspring of obese mothers have a latent defect in lipid and glucose metabolism that is unmasked by an energy-dense Western-style diet.
Maternal obesity and diabetes alters hepatic gene expression in male offspring
The phenotypic defects in ObC and ObHF mice cannot have a genetic basis, since the mice are genetically identical to LnC and LnHF mice. In order to obtain a more detailed picture of the defects induced by maternal obesity and diabetes, we assessed hepatic gene expression in 16 mice (4 from each group) using Affymetrix GeneChip Mouse Gene 1.0 ST arrays, which provide whole transcript coverage of 28,853 genes in the mouse genome. Using the statistical packages MessinaCitation24 and LimmaGP (see Materials and Methods), we identified 45 transcripts (42 unique genes) whose expression was significantly altered by maternal obesity (ObC vs. LnC; ). Candidate gene expression changes identified by LimmaGP were, as expected, concordant across all animals within a group (because it uses a moderated t-statistic), whereas changes identified by Messina had larger effect sizes but more heterogeneity within groups (because it allows for intra-group variation)Citation24; Messina candidates were also more likely to be associated with a CpG island (). qPCR was used to interrogate five candidate genes (chosen at random) associated with CpG islands, four of which exhibited altered expression in ObC relative to LnC as predicted by the array (Dbp, Gm129, Gna14, Zbtb16; Fig. S3). Validation included many animals that were not used on the array (17/22), and the same intragroup variation was observed in this larger cohort, particularly in the ObC group; this is consistent with a stochastic epigenetic response to maternal obesity.
Figure 3. The latent metabolic phenotype is associated with changes in hepatic gene expression. (A) Heat map of gene transcripts with significant expression changes in ObC vs. LnC offspring (n = 4 per group); note the similar patterning across all animals exposed to overnutrition (prenatally, postnatally or both). The columns on the right indicate genes that have CpG island promoters (green) or do not (white), and whether the gene was identified by LimmaGP (white), Messina (black) or both (gray). Average log2-fold expression changes are shown at the far right. (B) Bar graph showing –log p-values (with Benjamini–Hochberg correction for multiple testing) for gene ontologies significantly overrepresented in genes that are differentially expressed in ObC animals relative to LnC. A p-value cutoff of 0.05 is indicated by the red line. (C) Bar graph showing relative copy number of mitochondrial genes Atp6 and Nd6 (relative to the nuclear gene Ndufv1) in livers of LnC, ObC, LnHF and ObHF animals. Data are represented as mean ± SEM. Statistically significant differences are indicated by asterisks: *p < 0.05, **p < 0.01, ***p < 0.001.
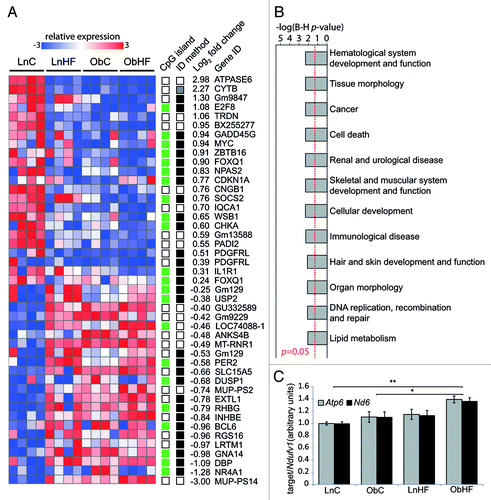
A similar pattern of expression changes was found in control mice exposed to the Western diet (LnHF vs. LnC); using the same array analysis parameters we find that 30 of the 45 transcripts (67%) identified as altered in ObC are also significantly altered in LnHF. This suggests that intrauterine and preweaning exposure to maternal obesity induces expression changes in a set of genes that are also responsive to overnutrition in later life. A much larger number of genes exhibited altered expression in the ObHF group compared with the other groups (Fig. S4); this is likely to reflect the obvious liver pathology in the ObHF mice (see ), which confounds any attempt to link gene expression changes to the latent predisposition in this group.
Gene ontology (GO) analysis of genes whose expression was altered in ObC as compared with LnC obtained a large set of ontologies, within which metabolic functions were present but not prominent (). The two genes with the most strongly downregulated expression were ATPASE6 and CYTB, both encoded by the mitochondrial genome. We asked if this decrease reflected a lower mitochondrial copy number in hepatocytes of ObC mice, but found no difference in mitochondrial genome copy number between LnC and ObC (). Among the most strongly upregulated genes are major urinary proteins (MUPs), which have been implicated in regulation of energy expenditure.Citation25
Maternal obesity causes widespread epigenetic changes in male offspring
The characteristics of the mice exposed to maternal obesity and diabetes demonstrate that they are programmed in fetal life to respond to an energy-dense diet by developing adiposity, insulin resistance, glucose intolerance and hepatic steatosis. This programming may involve stable alterations in gene expression states, mediated by epigenetic modifications. The molecular basis of epigenetic gene regulation is complex and incompletely characterized; however the role of cytosine methylation has been extensively investigated and is best understood in the suppression of transcription initiation.Citation26 Cytosine methylation in mammals is almost completely confined to CpG dinucleotides, which are concentrated in “CpG islands” that are transcriptional regulatory elements.
We compared the genome wide patterns of CpG island methylation in the livers of ObC and LnC mice (high fat-fed mice were not examined, as their overt metabolic disease would make it impossible to distinguish between methylation changes that were potentially causal of the latent phenotype, and those merely consequential to their disease). As previously described,Citation8 we made libraries enriched for the unmethylated fraction of DNA by sequential digestion with HpaII and McrBC, followed by ligation-mediated PCR. Libraries were hybridized to Agilent Mouse CpG Island 105K arrays covering approximately 16,000 CpG islands. We analyzed 8 male LnC mice and 8 male ObC mice; pooled libraries from 10 LnC controls acted as the reference sample for each array.
We first used principal components analysis (PCA) to visualize the overall distribution of array data. PCA reduces many variables within a complex data set to a few artificial variables, which together account for most of the variance in the data. The first four principal components of our data accounted for over half (54.4%) of the variability and are shown in two-dimensional score plots (). The intermingling of ObC and LnC mice in principal components (PC) 1 and 2 indicates that most variation is common within these two groups. This spontaneous inter-individual variation is reminiscent of the variation we have previously described in C57BL/6J mice.Citation8 However, distinct clustering and separation of ObC and LnC offspring is evident in PC 3 and 4. Taken together, the intermingling in PC 1 and 2 and separate clustering in PC 3 and 4 indicate that intergroup variation resides within a larger context of the spontaneous variation we and othersCitation8,Citation10 have previously described. A GO analysis of the 5% most methylation-variable loci in LnC and ObC, which are responsible for PC 1 and 2, produces a set of ontologies related to developmental functions () that are nearly identical to the set identified in our previous study of intrinsic epigenetic variation in the C57BL/6J strain.Citation8
Figure 4. Livers of obese-born males exhibit widespread changes in cytosine methylation. (A) Two-dimensional plots showing principal component (PC) scores of LnC and ObC animals (n = 8 per group). Three plots are shown: scores for PC 1 vs. PC2 (left), PC2 vs. PC 3 (middle) and PC 3 vs. PC 4 (right). The amount of variability accounted for by each PC is indicated on the axes. The wireframe ellipses surrounding PC scores for each group are indicative of the overall standard deviation within each group. (B) Bar graph showing –log p-values (with Benjamini–Hochberg correction for multiple testing) for gene ontologies significantly overrepresented in methylation variable genes in LnC and ObC animals. A p-value cutoff of 0.05 is indicated by the red dashed line. (C) Histogram showing the distribution of LimmaGP p-values across all probes on the methylation array. A p-value cutoff of 0.01 is indicated by the red dashed line. (D) Histogram showing the distribution of Messina margin scores from the methylation array. Inset shows enlarged view of the histogram tails. (E) Bar graph showing –log p-values (with Benjamini-Hochberg correction for multiple testing) for gene ontologies identified as being significantly overrepresented in genes differentially methylated in ObC vs LnC (as detected by LimmaGP, p < 0.01). A p-value cutoff of 0.05 is indicated by the red dashed line.
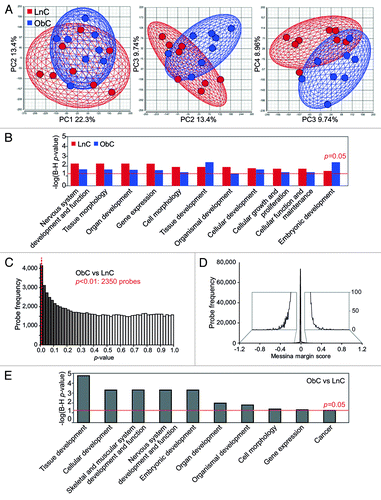
We investigated the array probes responsible for the separate clustering of ObC and LnC in PC 3 and 4. Statistical testing by LimmaGP revealed an enrichment of small p-values (), indicating that there are significant methylation differences between the two groups. Limma detected methylation differences at 2350 probes corresponding to 1598 genes (at p < 0.01). Given the substantial intragroup variation, we also used Messina to identify stochastic changes in methylation that affected most but not all individuals in the ObC group. Most margin scores were very small (); only 36 loci showed a margin of >1.5-fold change. The many more significant methylation changes identified by LimmaGP () are therefore likely to be small in magnitude. Validation by bisulfite allelic sequencing showed that 6 of 9 loci examined exhibited small methylation differences (0.9–15.3%, Fig. S5) in ObC animals in the direction predicted by the array. The verification rate (FDR ~0.33) and the small magnitude of the changes are consistent with that of other studies of complex phenotypes using genome-wide strategies.Citation8,Citation27-Citation30 There was no significant overlap between the genes whose expression was changed in the ObC group and the genes whose methylation states differed. However, GO analysis of the genes identified by LimmaGP as differentially methylated (at p < 0.01) between the ObC and LnC groups again revealed significant enrichment in ontologies associated with development (); these are largely the same ontologies (8/10) as those in the intrinsically methylation-variable loci (). Despite the prominent metabolic phenotype in Ob mice, metabolic functions are not represented in these ontologies. This may suggest that the effects of maternal obesity are much broader than the metabolic aberrations uncovered by a Western diet.
Discussion
We have found that natural-onset maternal obesity and diabetes induces a latent defect in glucose and lipid metabolism in offspring of Avy mice. This defect is unmasked by a Western-style diet, which causes mice to become insulin resistant and develop hepatic steatosis. Changes in gene expression induced by maternal obesity point to a broad set of functional ontologies, not limited to energy metabolism. Genome-wide assay of CpG island methylation demonstrates that maternal obesity induces widespread methylation changes, which affect functional ontologies relating to development and gene expression. Thus the latent defect in energy metabolism is not prominently reflected in the expression and epigenomic changes induced by maternal obesity.
The latent metabolic phenotype we observed in this study contrasts with those of other rodent models using high fat and sugar diets to mimic maternal overnutrition: in these cases glucose handling defects are apparent in offspring even when they are weaned onto a normal chow diet.Citation21,Citation31 The syndrome of obese yellow Avy/a mice more closely models the gradual onset and progression of obesity and type 2 diabetes in humans. In this model, a/a offspring exhibit only subtle defects in lipid metabolism and do not progress to a disease state if they are maintained on a standard chow diet. Exposure to an energy-dense “Western” diet produces overt effects, which result in mice exhibiting the equivalent of nonalcoholic steatohepatitis (NASH) in humans. If this effect occurs in humans, it would suggest that the children of obese and diabetic mothers are at risk of metabolic syndrome but can be protected by avoiding an energy-dense diet.
Fetal programming by maternal nutrition has been proposed as a predictive-adaptive response to environmental conditions, whereby developing offspring adjust their physiology to suit the expected postnatal environment. While the underlying mechanisms are not clear, it is now generally supposed that epigenetic modification of metabolically relevant genes is involved.Citation32 But concrete examples are limited to a handful of genes, where the observed changes are small,Citation33-Citation36 and very few exist in the context of maternal overnutrition.Citation37,Citation38 Here we deliberately took an unbiased, genome-wide approach to ask whether epigenetic mechanisms are involved in the response to maternal obesity. While we found that maternal obesity induced methylation changes across the genome and expression changes at a variety of genes, metabolic genes were notable by their relative absence. Instead, the genes whose methylation is affected by maternal obesity cluster in developmental ontologies. This may indicate a greater susceptibility of developmental processes to environmental perturbation, consistent with our previous finding that methylation patterns at genes in developmental ontologies are intrinsically more variable and susceptible to dietary supplements than other parts of the genome.Citation8 The systemic consequences of a perturbation in developmental processes could well underlie the metabolic phenotype and perhaps other cryptic phenotypes programmed by maternal obesity and diabetes. Recent large-scale epidemiological studies on the prevalence of non-metabolic disorders, such as asthma and autism in children of obese mothers, support the idea that other defects may be programmed by maternal obesity.Citation39,Citation40
The methylation changes we observed were small in magnitude, consistent with previous reports of epigenetic programming by us and others.Citation8,Citation11,Citation36,Citation41,Citation42 This raises the obvious question as to the relevance of the methylation changes to the phenotype. Certainly, we did not observe a large overlap between the genes with methylation changes and those with changed expression. This is reminiscent of the findings of Rando and colleagues, where widespread methylation changes in response to paternal high-fat feeding did not specify individual gene expression changes, but were nevertheless linked to a metabolic phenotype in offspring.Citation41 Notably, few metabolic genes were altered in this study, and those that were showed a magnitude of methylation change similar to that we observe here.
The latency of the metabolic phenotype in our model permits the examination of methylation and gene expression changes that occur prior to the onset of disease, allowing us to avoid detecting changes that are consequential to disease. Consistent with their metabolic phenotype, the high-fat fed group exhibited gene expression changes related to metabolism; this is a common finding in fetal programming studies, and such changes are often assumed to be inborn. But in our control fed offspring, where disease is not yet present, we find that metabolic genes are practically absent from comparisons between lean- and obese-born mice. The possible exceptions are the mitochondrially-encoded genes involved in oxidoreductase activity: mitochondrial activity is known to play an important role in the pathogenesis of metabolic disease and particularly, in NASH.Citation43 We speculate that the changes we observe in expression of genes related to non-metabolic functional ontologies may either play an indirect role in regulating metabolism, or indicate other cryptic defects in these mice, programmed by maternal obesity but possibly unrelated to energy metabolism.
The predictive-adaptive view of fetal programming has been heavily influenced by the idea that the infants of undernourished mothers are adapted to an environment with scarce food resources.Citation1 An alternative possibility is that inappropriate nutrition (undernutrition, overnutrition or a modified supply of key nutrients) programs generalized defects that impair responses to environmental stresses or conditions. In this view, the relative paucity of metabolic genes in our lists of affected loci is not surprising. Since diet is always likely to be among the most prominent environmental conditions, defects related to the handling of nutrients may be the most readily unmasked; in our study this seems to particularly be the case with male offspring. Sexually dimorphic responses have been observed in a number of fetal programming studies (reviewed in ref. Citation44); while the effect of sex hormones is often invoked as the mediator of sexual dimorphism in the setting of metabolic programming, it may also be that sex-specific epigenetic profiles render either the male or female genomes more vulnerable to the effects of specific environmental insults,Citation45 either in utero or postnatally.
The fetal response to maternal obesity is clearly maladaptive and the epigenomic response most likely represents some form of damage induced by a suboptimal intrauterine environment. However, the maladapted phenotype is not a fait accompli: When maintained on a healthy diet, the mice never develop metabolic disease. Thus, although risk is programmed, overt disease is avoidable; if this translates to human populations, it highlights the need for multigenerational affirmative intervention to break the cycle of obesity.
Materials and Methods
Mouse breeding and diets
All animals were handled in accordance with good practice as defined by the National Health and Medical Research Council (Australia) Statement on Animal Experimentation and requirements of state government legislation. The study was approved by the Garvan/St. Vincent’s Animal Ethics Committee (06/12 and 09/12).
The Avy mice used in this study were descended from an isogenic C57BL/6 colony at Oak Ridge National Laboratories and have been maintained at the Victor Chang Cardiac Research Institute since 2001. All parental mice were fed ad libitum on NIH-31 control diet (5% w/w fat, 13.5 MJ/kg). a/a offspring were weaned onto either NIH-31 or SF00-219 high-fat diet designed to mimic a Western fast-food diet [equivalent to Harlan Teklad TD88137; 22% w/w fat (40% digestible energy), 0.15% w/w cholesterol, 19.4 MJ/kg]; Avy/a littermates were culled at weaning. Feeds were manufactured by Specialty Feeds.
Glucose and insulin tolerance tests
For metabolic testing, mice were fasted for 6 h and given an intraperitoneal injection of either 1.5 g glucose/kg mouse weight (for glucose tolerance test) or 0.75 units of insulin/kg mouse weight (for insulin tolerance test). Blood glucose levels were then measured at 15 min (glucose) or 10 min (insulin) intervals using an Accu-Chek II Performa® glucose monitor (Roche Diagnostics GmbH).
Lipid profiling
Lipidomic profiling was performed on liver samples as described previously.Citation46
Liver histology
Haematoxylin and eosin stained sections were scored by an anatomical pathologist (M.E.B.) for nonalcoholic steatohepatitis according to standardized pathological criteria.Citation22
Mitochondrial copy number
Mitochondrial copy number was assessed by qPCR quantification of mitochondrial genes Atp6 and Nd6 normalized to the nuclear gene Ndufv1.Citation47 Each reaction was done in triplicate with approximately 10 ng of DNA per reaction. See Table S1 for primer sequences.
Leptin assay
Serum was collected at 12 weeks, and leptin concentrations were measured using the Mouse Leptin ELISA (Millipore) following manufacturer’s instructions.
Statistical analysis for physiological data
All comparisons were made between age-matched, gender-matched, genetically identical mice. Statistical significance was determined by two-way ANOVA with two-tailed Student’s t-tests for glucose and insulin tolerance tests, by two-tailed Student’s t-tests for the other metabolic tests and by a right-tailed Fisher’s exact test for comparison of steatosis scores.
Gene expression
Total RNA was extracted from liver tissue and analyzed with GeneChip® Mouse Gene 1.0ST arrays (Affymetrix). Labeling and hybridization were performed by the Ramaciotti Centre for Gene Function Analysis (University of New South Wales). Microarray data was preprocessed and normalized using robust multichip average (RMA), via the NormalizeAffymetrixST (version 2.0, available at pwbc.garvan.unsw.edu.au/gp) GenePattern module (version 3.2.348). Differential gene expression analysis was performed using LimmaGP and Messina (see below).
LimmaGP analysis
LimmaGP (version 19.3, available at pwbc.garvan.unsw.edu.au/gp; Cowley et al., manuscript in preparation) is a GenePattern module for identifying differential gene expression using limma.Citation49 Briefly, limma combines linear models to flexibly represent the experimental design of a microarray study in a mathematical framework, with an empirical Bayes, moderated t-statistic, which has more power than the Student’s t-test or ANOVA for analyzing microarray data.Citation49 We chose a cutoff of FDR <0.05, with p < 1 × 10−5 and a minimum fold change of 1.3, to capture genes with a consistent change between groups.
Messina analysis
We used MessinaCitation24 as a complimentary candidate identification method. Messina takes a machine learning approach and allows for intra-group heterogeneity by permitting a user-specified, low level of misclassification. This allows detection of candidate genes with inconsistent changes in expression, so is particularly suited to studies where changes may be stochastic. Messina represents the magnitude of differential expression by a margin score (representing the difference in expression between the closest included members of the two groups). We set a maximum misclassification of one animal out of four in both control and test groups (i.e., specificity and sensitivity equals 0.75), and applied a margin of ≥0.585 (equivalent to ≥1.5-fold change).
Microarray validation
One microgram total RNA was reverse transcribed and assayed by qPCR. Each reaction was done in triplicate with approximately 10 ng of cDNA per reaction. Samples were normalized to the geometric mean of four housekeeping genes (Actb, Hprt, Gapdh and Canx); see Table S1 for primer sequences.
Methylation analysis
DNA was extracted, enriched for unmethylated sequence by HpaII and McrBC digest, and hybridized to two-color Agilent Mouse 105K CpG island arrays as previously described.Citation8 Each sample was hybridized against a reference pool made by combining DNA libraries from 10 LnC offspring. Data was analyzed with LimmaGP and Messina algorithms as described above. Principal components analysis was performed in Partek Genomics Suite (v6.5).
Bisulfite sequencing validation
Allelic methylation patterns at nine candidate loci were assessed by bisulfite allelic sequencing.Citation50 Two micrograms DNA was treated with sodium bisulfite using the Epitect kit (QIAGEN GmbH), PCR amplified, cloned into E.Coli and sequenced. See Table S1 for primer sequences.
Gene ontology
Gene ontology analysis was performed on gene expression and methylation candidates using the standard workflow in Ingenuity Pathways Analysis (Ingenuity Systems).
| Abbreviations: | ||
| Avy | = | agouti viable yellow |
Additional material
Download Zip (14.3 MB)Acknowlegdements
The authors gratefully acknowledge the technical assistance of Stephanie Hackworthy, Natalie Stunnell and Mark Pinese at VCCRI, the Ramaciotti Centre at the University of New South Wales and Jacqueline Weir and Associate Professor Peter Meikle at Baker IDI. This study was supported by the National Health and Medical Research Council (NHMRC) of Australia (ID514911 and ID635510). C.M.S. is an Australian Research Council (ARC) Future Fellow. J.E.C. is an ARC DECRA Fellow. M.A.F. is a Senior Principal Research Fellow of the NHMRC. D.C.H. is supported by a National Heart Foundation (NHF) Biomedical Postdoctoral Fellowship.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplementary Material
Supplementary materials may be found here:
http://www.landesbioscience.com/journals/epigenetics/article/24656
Notes
† These authors contributed equally to this work.
References
- Alfaradhi MZ, Ozanne SE. Developmental programming in response to maternal overnutrition. Front Genet 2011; 2:27; http://dx.doi.org/10.3389/fgene.2011.00027; PMID: 22303323
- McMillen IC, Robinson JS. Developmental origins of the metabolic syndrome: prediction, plasticity, and programming. Physiol Rev 2005; 85:571 - 633; http://dx.doi.org/10.1152/physrev.00053.2003; PMID: 15788706
- Armitage JA, Taylor PD, Poston L. Experimental models of developmental programming: consequences of exposure to an energy rich diet during development. J Physiol 2005; 565:3 - 8; http://dx.doi.org/10.1113/jphysiol.2004.079756; PMID: 15695245
- Jawerbaum A, White V. Animal models in diabetes and pregnancy. Endocr Rev 2010; 31:680 - 701; http://dx.doi.org/10.1210/er.2009-0038; PMID: 20534704
- Carmody JS, Wan P, Accili D, Zeltser LM, Leibel RL. Respective contributions of maternal insulin resistance and diet to metabolic and hypothalamic phenotypes of progeny. Obesity (Silver Spring) 2011; 19:492 - 9; http://dx.doi.org/10.1038/oby.2010.245; PMID: 20948526
- White CL, Purpera MN, Morrison CD. Maternal obesity is necessary for programming effect of high-fat diet on offspring. Am J Physiol Regul Integr Comp Physiol 2009; 296:R1464 - 72; http://dx.doi.org/10.1152/ajpregu.91015.2008; PMID: 19244583
- Gallou-Kabani C, Junien C. Nutritional epigenomics of metabolic syndrome: new perspective against the epidemic. Diabetes 2005; 54:1899 - 906; http://dx.doi.org/10.2337/diabetes.54.7.1899; PMID: 15983188
- Li CCY, Cropley JE, Cowley MJ, Preiss T, Martin DIK, Suter CM. A sustained dietary change increases epigenetic variation in isogenic mice. PLoS Genet 2011; 7:e1001380; http://dx.doi.org/10.1371/journal.pgen.1001380; PMID: 21541011
- Bird A. DNA methylation patterns and epigenetic memory. Genes Dev 2002; 16:6 - 21; http://dx.doi.org/10.1101/gad.947102; PMID: 11782440
- Feinberg AP, Irizarry RA. Evolution in health and medicine Sackler colloquium: Stochastic epigenetic variation as a driving force of development, evolutionary adaptation, and disease. Proc Natl Acad Sci U S A 2010; 107:Suppl 1 1757 - 64; http://dx.doi.org/10.1073/pnas.0906183107; PMID: 20080672
- Feinberg AP, Irizarry RA, Fradin D, Aryee MJ, Murakami P, Aspelund T, et al. Personalized epigenomic signatures that are stable over time and covary with body mass index. Sci Transl Med 2010; 2:49ra67; http://dx.doi.org/10.1126/scitranslmed.3001262; PMID: 20844285
- Wolff GL. Body Composition and Coat Color Correlation in Different Phenotypes of “Viable Yellow” Mice. Science 1965; 147:1145 - 7; http://dx.doi.org/10.1126/science.147.3662.1145; PMID: 14242032
- Yen TT, Gill AM, Frigeri LG, Barsh GS, Wolff GL. Obesity, diabetes, and neoplasia in yellow A(vy)/- mice: ectopic expression of the agouti gene. FASEB J 1994; 8:479 - 88; PMID: 8181666
- Duhl DM, Vrieling H, Miller KA, Wolff GL, Barsh GS. Neomorphic agouti mutations in obese yellow mice. Nat Genet 1994; 8:59 - 65; http://dx.doi.org/10.1038/ng0994-59; PMID: 7987393
- Frigeri LG, Wolff GL, Teguh C. Differential responses of yellow Avy/A and agouti A/a (BALB/c X VY) F1 hybrid mice to the same diets: glucose tolerance, weight gain, and adipocyte cellularity. Int J Obes 1988; 12:305 - 20; PMID: 3058616
- Yen TT, Allan JA, Yu PL, Acton MA, Pearson DV. Triacylglycerol contents and in vivo lipogenesis of ob/ob, db/db and Avy/a mice. Biochim Biophys Acta 1976; 441:213 - 20; http://dx.doi.org/10.1016/0005-2760(76)90164-8; PMID: 952988
- Wolff GL, Roberts DW, Mountjoy KG. Physiological consequences of ectopic agouti gene expression: the yellow obese mouse syndrome. Physiol Genomics 1999; 1:151 - 63; PMID: 11015573
- Kennedy AJ, Ellacott KL, King VL, Hasty AH. Mouse models of the metabolic syndrome. Dis Model Mech 2010; 3:156 - 66; http://dx.doi.org/10.1242/dmm.003467; PMID: 20212084
- Samuelsson AM, Matthews PA, Argenton M, Christie MR, McConnell JM, Jansen EH, et al. Diet-induced obesity in female mice leads to offspring hyperphagia, adiposity, hypertension, and insulin resistance: a novel murine model of developmental programming. Hypertension 2008; 51:383 - 92; http://dx.doi.org/10.1161/HYPERTENSIONAHA.107.101477; PMID: 18086952
- Nivoit P, Morens C, Van Assche FA, Jansen E, Poston L, Remacle C, et al. Established diet-induced obesity in female rats leads to offspring hyperphagia, adiposity and insulin resistance. Diabetologia 2009; 52:1133 - 42; http://dx.doi.org/10.1007/s00125-009-1316-9; PMID: 19288075
- Taylor PD, McConnell J, Khan IY, Holemans K, Lawrence KM, Asare-Anane H, et al. Impaired glucose homeostasis and mitochondrial abnormalities in offspring of rats fed a fat-rich diet in pregnancy. Am J Physiol Regul Integr Comp Physiol 2005; 288:R134 - 9; http://dx.doi.org/10.1152/ajpregu.00355.2004; PMID: 15388492
- Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, et al, Nonalcoholic Steatohepatitis Clinical Research Network. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005; 41:1313 - 21; http://dx.doi.org/10.1002/hep.20701; PMID: 15915461
- Maffei M, Halaas J, Ravussin E, Pratley RE, Lee GH, Zhang Y, et al. Leptin levels in human and rodent: measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nat Med 1995; 1:1155 - 61; http://dx.doi.org/10.1038/nm1195-1155; PMID: 7584987
- Pinese M, Scarlett CJ, Kench JG, Colvin EK, Segara D, Henshall SM, et al. Messina: a novel analysis tool to identify biologically relevant molecules in disease. PLoS One 2009; 4:e5337; http://dx.doi.org/10.1371/journal.pone.0005337; PMID: 19399185
- Zhou Y, Rui L. Major urinary protein regulation of chemical communication and nutrient metabolism. Vitam Horm 2010; 83:151 - 63; http://dx.doi.org/10.1016/S0083-6729(10)83006-7; PMID: 20831945
- Deaton AM, Bird A. CpG islands and the regulation of transcription. Genes Dev 2011; 25:1010 - 22; http://dx.doi.org/10.1101/gad.2037511; PMID: 21576262
- Flanagan JM, Popendikyte V, Pozdniakovaite N, Sobolev M, Assadzadeh A, Schumacher A, et al. Intra- and interindividual epigenetic variation in human germ cells. Am J Hum Genet 2006; 79:67 - 84; http://dx.doi.org/10.1086/504729; PMID: 16773567
- Mill J, Tang T, Kaminsky Z, Khare T, Yazdanpanah S, Bouchard L, et al. Epigenomic profiling reveals DNA-methylation changes associated with major psychosis. Am J Hum Genet 2008; 82:696 - 711; http://dx.doi.org/10.1016/j.ajhg.2008.01.008; PMID: 18319075
- Rakyan VK, Beyan H, Down TA, Hawa MI, Maslau S, Aden D, et al. Identification of type 1 diabetes-associated DNA methylation variable positions that precede disease diagnosis. PLoS Genet 2011; 7:e1002300; http://dx.doi.org/10.1371/journal.pgen.1002300; PMID: 21980303
- Einstein F, Thompson RF, Bhagat TD, Fazzari MJ, Verma A, Barzilai N, et al. Cytosine methylation dysregulation in neonates following intrauterine growth restriction. PLoS One 2010; 5:e8887; http://dx.doi.org/10.1371/journal.pone.0008887; PMID: 20126273
- Srinivasan M, Katewa SD, Palaniyappan A, Pandya JD, Patel MS. Maternal high-fat diet consumption results in fetal malprogramming predisposing to the onset of metabolic syndrome-like phenotype in adulthood. Am J Physiol Endocrinol Metab 2006; 291:E792 - 9; http://dx.doi.org/10.1152/ajpendo.00078.2006; PMID: 16720630
- Gluckman PD, Hanson MA, Buklijas T, Low FM, Beedle AS. Epigenetic mechanisms that underpin metabolic and cardiovascular diseases. Nat Rev Endocrinol 2009; 5:401 - 8; http://dx.doi.org/10.1038/nrendo.2009.102; PMID: 19488075
- Lillycrop KA, Phillips ES, Jackson AA, Hanson MA, Burdge GC. Dietary protein restriction of pregnant rats induces and folic acid supplementation prevents epigenetic modification of hepatic gene expression in the offspring. J Nutr 2005; 135:1382 - 6; PMID: 15930441
- Park JH, Stoffers DA, Nicholls RD, Simmons RA. Development of type 2 diabetes following intrauterine growth retardation in rats is associated with progressive epigenetic silencing of Pdx1. J Clin Invest 2008; 118:2316 - 24; PMID: 18464933
- Bogdarina I, Welham S, King PJ, Burns SP, Clark AJ. Epigenetic modification of the renin-angiotensin system in the fetal programming of hypertension. Circ Res 2007; 100:520 - 6; http://dx.doi.org/10.1161/01.RES.0000258855.60637.58; PMID: 17255528
- Heijmans BT, Tobi EW, Stein AD, Putter H, Blauw GJ, Susser ES, et al. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc Natl Acad Sci U S A 2008; 105:17046 - 9; http://dx.doi.org/10.1073/pnas.0806560105; PMID: 18955703
- El Hajj N, Pliushch G, Schneider E, Dittrich M, Muller T, Korenkov M, et al. Metabolic programming of MEST DNA methylation by intrauterine exposure to gestational diabetes mellitus. Diabetes 2013; 62:1320 - 8; http://dx.doi.org/10.2337/db12-0289; PMID: 23209187
- Gemma C, Sookoian S, Alvariñas J, García SI, Quintana L, Kanevsky D, et al. Maternal pregestational BMI is associated with methylation of the PPARGC1A promoter in newborns. Obesity (Silver Spring) 2009; 17:1032 - 9; http://dx.doi.org/10.1038/oby.2008.605; PMID: 19148128
- Krakowiak P, Walker CK, Bremer AA, Baker AS, Ozonoff S, Hansen RL, et al. Maternal metabolic conditions and risk for autism and other neurodevelopmental disorders. Pediatrics 2012; 129:e1121 - 8; http://dx.doi.org/10.1542/peds.2011-2583; PMID: 22492772
- Lowe A, Bråbäck L, Ekeus C, Hjern A, Forsberg B. Maternal obesity during pregnancy as a risk for early-life asthma. J Allergy Clin Immunol 2011; 128:1107 - 9, e1-2; http://dx.doi.org/10.1016/j.jaci.2011.08.025; PMID: 21958587
- Carone BR, Fauquier L, Habib N, Shea JM, Hart CE, Li R, et al. Paternally induced transgenerational environmental reprogramming of metabolic gene expression in mammals. Cell 2010; 143:1084 - 96; http://dx.doi.org/10.1016/j.cell.2010.12.008; PMID: 21183072
- Ng S-F, Lin RC, Laybutt DR, Barres R, Owens JA, Morris MJ. Chronic high-fat diet in fathers programs β-cell dysfunction in female rat offspring. Nature 2010; 467:963 - 6; http://dx.doi.org/10.1038/nature09491; PMID: 20962845
- Szendroedi J, Phielix E, Roden M. The role of mitochondria in insulin resistance and type 2 diabetes mellitus. Nat Rev Endocrinol 2012; 8:92 - 103; http://dx.doi.org/10.1038/nrendo.2011.138; PMID: 21912398
- Hochberg Z, Feil R, Constancia M, Fraga M, Junien C, Carel JC, et al. Child health, developmental plasticity, and epigenetic programming. Endocr Rev 2011; 32:159 - 224; http://dx.doi.org/10.1210/er.2009-0039; PMID: 20971919
- Gabory A, Attig L, Junien C. Sexual dimorphism in environmental epigenetic programming. Mol Cell Endocrinol 2009; 304:8 - 18; http://dx.doi.org/10.1016/j.mce.2009.02.015; PMID: 19433243
- Matthews VB, Allen TL, Risis S, Chan MH, Henstridge DC, Watson N, et al. Interleukin-6-deficient mice develop hepatic inflammation and systemic insulin resistance. Diabetologia 2010; 53:2431 - 41; http://dx.doi.org/10.1007/s00125-010-1865-y; PMID: 20697689
- Guo W, Jiang L, Bhasin S, Khan SM, Swerdlow RH. DNA extraction procedures meaningfully influence qPCR-based mtDNA copy number determination. Mitochondrion 2009; 9:261 - 5; http://dx.doi.org/10.1016/j.mito.2009.03.003; PMID: 19324101
- Reich M, Liefeld T, Gould J, Lerner J, Tamayo P, Mesirov JP. GenePattern 2.0. Nat Genet 2006; 38:500 - 1; http://dx.doi.org/10.1038/ng0506-500; PMID: 16642009
- Smyth GK. Limma: linear models for microarray data. In: Gentleman R, Carey V, Dudoit S, Irizarry RA, Huber W, eds. Bioinformatics and computational biology solutions using r and bioconductor. New York: Springer, 2005:397-420.
- Clark SJ, Harrison J, Paul CL, Frommer M. High sensitivity mapping of methylated cytosines. Nucleic Acids Res 1994; 22:2990 - 7; http://dx.doi.org/10.1093/nar/22.15.2990; PMID: 8065911