Abstract
CpG Island Methylator Phenotype (CIMP) is one of the underlying mechanisms in colorectal cancer (CRC). This study aimed to define a methylome signature in CRC through a methylation microarray analysis and a compilation of promising CIMP markers from the literature. Illumina HumanMethylation27 (IHM27) array data was generated and analyzed based on statistical differences in methylation data (1st approach) or based on overall differences in methylation percentages using lower 95% CI (2nd approach). Pyrosequencing was performed for the validation of nine genes. A meta-analysis was used to identify CIMP and non-CIMP markers that were hypermethylated in CRC but did not yet make it to the CIMP genes’ list. Our 1st approach for array data analysis demonstrated the limitations in selecting genes for further validation, highlighting the need for the 2nd bioinformatics approach to adequately select genes with differential aberrant methylation. A more comprehensive list, which included non-CIMP genes, such as APC, EVL, CD109, PTEN, TWIST1, DCC, PTPRD, SFRP1, ICAM5, RASSF1A, EYA4, 30ST2, LAMA1, KCNQ5, ADHEF1, and TFPI2, was established. Array data are useful to categorize and cluster colonic lesions based on their global methylation profiles; however, its usefulness in identifying robust methylation markers is limited and rely on the data analysis method. We have identified 16 non-CIMP-panel genes for which we provide rationale for inclusion in a more comprehensive characterization of CIMP+ CRCs. The identification of a definitive list for methylome specific genes in CRC will contribute to better clinical management of CRC patients.
Keywords: :
Introduction
Recently, we have witnessed a surge of interest in epigenetics and recent advances in the field have revolutionized our understanding of various biological processes and complex diseases, most notably colorectal cancer (CRC). Changes that transform normal tissue to CRC occur via multistep genetic and epigenetic alterations that include DNA methylation [CpG Methylator Phenotype (CIMP)], histone modifications and miRNAs. Studies revealed that most critical processes found in cancer cells can be triggered directly or indirectly by epigenetic deregulation.
CIMP gene-panels have differed among different research laboratories, and there is a lack of consensus on the ideal list of CIMP markers.Citation1 In a previous study, hierarchical clustering was performed in CRCs for the identification of two panels of methylation markers based on the epigenotypes: high-, intermediate-, and low-methylation (HME, IME, and LME, respectively).Citation2 It is important to note that the CIMP gene-panels significantly correlate with mutations in BRAF and KRAS genes, in addition to the microsatellite instability (MSI) status of CRCs. Indeed, markers such as MLH1 and p16 were found to be highly methylated in MSI CRC tumors. Although several CIMP-related genes have already been described in the literature, new studies indicate the existence of other potential genes that are frequently methylated, but have thus far not been recognized as CIMP-markers.
For instance, APC gene expression is inhibited through mutation and promoter hypermethylation.Citation3-Citation5 Additionally, APC is a CRC progression marker,Citation6 and its expression maybe downregulated in precancerous adenomatous tissue via promoter hypermethylation.Citation7 Likewise, 3OST2 is highly methylated (82%) in CRC, including sporadic and Lynch Syndrome CRC.Citation8,Citation9 In another study investigating multiple and solitary tumors from 47 patients, more than 70% showed promoter hypermethylation.Citation10 The extracellular matrix (ECM) gene EVL is silenced by promoter methylation in > 50% of CRCs, and is a predictor of poor survival.Citation11-Citation13 An intronic region of the EVL gene contains a micro-RNA (hsa-miR-342) that is regulated by EVL promoter methylation. In addition to EVL expression itself, > 65% of adenomas and > 85% CRCs evaluated in a study showed hypermethylation of has-miR-342, suggesting that both EVL and has-miR-342 function as tumor suppressors in CRC.Citation14
Other genes such as PTEN,Citation15-Citation19 have been found to be frequently hypermethylated in CRC. TWIST1, which is a helix-loop-helix transcription factor regulated by the Wnt pathway has been shown to be hypermethylated in CRC in > 60% of tumor samples.Citation20-Citation22 Deleted in colon cancer (DCC) methylation is a frequent event in CRC, and three independent studies have found high rates (82%, 80%, and 56%) of DCC promoter methylation in primary and sporadic CRC using MSP, COBRA, and qMSP approaches, respectively.Citation23-Citation28 Genome-wide analysis have cited Receptor Protein Tyrosine Phosphatase Delta gene (PTPRD) as hypermethylated in CRC and a predictor of poor prognosis,Citation12 an observation that was previously corroborated by our laboratory in Iranian and African American populations.Citation13
Additional Wnt signaling pathway genes, such as Secreted Frizzled Related Protein 1 (SFRP1), are frequently hypermethylated in CRC,Citation29-Citation31 and high methylation of SFRP1 in normal rectal mucosa can be associated with aging, as per Worthley et al.Citation32,Citation33 Since SFRP1 can have high levels of methylation in normal colonic mucosa, and its promoter hypermethylation is convincingly involved in CRC pathogenesis, it will be important for future analysis to determine whether aberrant methylation of this gene is significantly different between cancer and normal adjacent tissue from individual CRC patients. RASSF1A, another Wnt pathway gene is often methylation-silenced in CRC.Citation16,Citation34-Citation39
The non-thiol protein tyrosine phosphatase EYA4 has recently been identified as a potential tumor suppressor gene in CRC using multiple detection techniques.Citation21,Citation40,Citation41,Citation42 Furthermore, it was reported that promoter of ECM pathway gene, Laminin α 1 (LAMA1), is methylated in nearly 67% of CRC patients tested.Citation43,Citation44 One of the 20 top ranking hypermethylated genes in CRC is Potassium voltage-gated channel, KQT-like subfamily, member 5 (KCNQ5).,,44 Similarly, the ADHFE1–Alcohol dehydrogenase, iron containing, 1 gene has the potential to be in the CIMP panel due to increased methylation levels in tumors compared with normal colorectal mucosa.Citation43,Citation44 Tissue Factor Pathway Inhibitor 2 (TFPI2), a potential tumor suppressor gene,Citation26 was shown to be methylated in > 97% adenomas and CRCs.Citation43,Citation45,Citation46 TFPI2 has been studied in a few independent ethnic groups,Citation25,Citation26,Citation45,Citation46 and its aberrant methylation was observed in all CRC studies.
In view of all the compelling data for the numerous potential genes that are currently not considered as CIMP genes, but definitely seem as likely candidates for CIMP, we conducted a microarray analysis of methylated genes in normal, adenoma and carcinoma from African American CRC patients and re-evaluated the status of CRC methylated genes with the intention to establish more robust CIMP gene-panel for better diagnosis and prognosis.
Results
Global methylation profiles and sample clustering
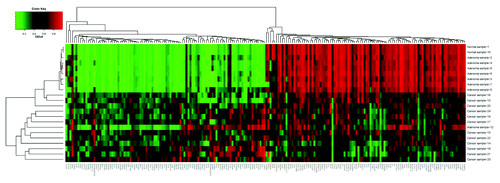
Using the IHM27 microarray, we generated methylation profiles of 12 cancers, 8 adenomas and 2 normal colon tissues. A clustering of these samples based on their methylome profiles led to a clear resolution between the cancer samples and the adenomas. The normal tissues clustered with the adenomas (). One adenoma clustered with the cancer samples. This adenoma was of a tubulovillous type, pointing to its advanced status in contrast to tubular adenomas.
Figure 1. Cluster profile analysis of colonic samples. Using the IHM27, we generated the methylation profiles of 12 cancers, 8 adenomas and 2 normal colonic tissues. A clustering of the different samples based on their methylome profiles led to a clear resolution between the cancer samples and the adenomas. The normal tissue clustered with the adenomas. With the exception of one adenoma (tubuvillus type) all the cancer samples was clustered together.
Bioinformatics approaches for IMH27 data analysis
Here we used two bioinformatics approaches for the identification of CIMP genes in our present study.
1st bioinformatics approach
Our initial approach for identifying genes to be validated using Illumina HumanMethylation27 gene array alone was unsuccessful (, left side of flowchart). Using this approach, 7,603 targets (28%) of the 27,578 CpG islands analyzed had a false discovery rate (FDR) adjusted P-value ≤ 0.002, and were selected as a reference point to determine the most significant CpG sites with aberrant methylation for validation. In the CRC group, 171 targets with β values ≥ 0.9 were considered as hypermethylated. Of these, 118 targets had β values ≤ 0.04 and were considered hypomethylated in the CRC group. To further identify targets for validation, the top 10% hypermethylated and the top 4% hypomethylated targets in the CRC group, were cross-referenced with 7,603 targets (FDR adjusted P ≤ 0.002).
Figure 2. Schematic representation of approaches used to identify appropriate genes for validation of CpG island methylation resulting from Illumina HumanMethylation27 Array data. The Illumina HumanMethylation27 Array analyzes 27,579 individual CpG sites and bioinformatics approaches are used to sort the data. The left side of the flowchart highlights our first approach at selecting genes for validation, which was unsuccessful. The right side of the flowchart highlights our second approach at selecting genes for validation, which is currently being attempted in the lab.
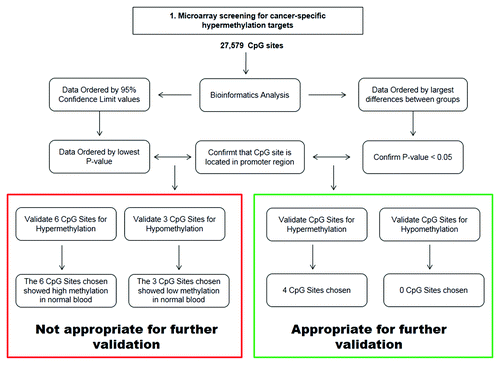
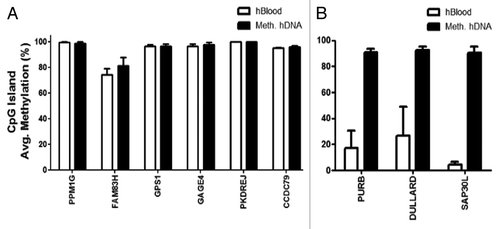
Six top performing CpG sites (PPM1G, FAM83H, GPS1, GAGE4, PKDREJ, and CCDC79; ) out of 171 were selected for subsequent validation of hypermethylation and 3 out of 118 CpG sites (PURB, DULLARD, and SAP30L) were validated for hypomethylation in CRC (). This bioinformatics approach appears to be inadequate for choosing CpG sites for validation. All 6 CpG sites that were found to be hypermethylated using this bioinformatics approach showed high levels of methylation in normal human blood (hBlood) samples as compared with the methylated human DNA control (Meth hDNA) (). Consistent with the hypermethylation analysis, the CpG sites analyzed for hypomethylation (PURB, DULLARD, and SAP30L) showed low methylation in hBlood compared with Meth hDNA (). These data suggested that this bioinformatics approach for choosing representative CpG sites for validation was inadequate and required further refinement (, red box).
Figure 3. Pyrosequencing validation of Illumina HumanMethylation27 array data. Pyrosequencing results of CpG islands that include the CpG site that was analyzed by the Illumina array for each gene. (A) Validation results of Illumina hypermethylated genes. (B) Validation results of Illumina hypomethylated genes. Error bars represent STD of methylation (%) between CpG sites within the CpG islands analyzed. No statistical differences were found.
2nd bioinformatics approach
The second approach for choosing genes for validation was based on finding the biggest differences between two groups that were part of the Illumina array cohort consisting of colorectal adenoma and cancer. After the bioinformatics analysis, the data was ordered from largest to smallest difference, where the differences were determined by subtracting the upper 95% confidence limits (u95CL) between groups. For hypermethylation in cancer vs. adenoma, the u95CL of adenoma cohorts was subtracted from those of cancers. After the data was ordered from largest to smallest differences, the next steps were to confirm that the FDR adjusted P-values are < 0.05, and determine if the CpG site analyzed by the array was within the promoter region of the gene (, right side of flowchart). Recently, the genomic coverage of the array was dramatically increased, leading to the production of the Infinium HumanMethylation450 (450K) BeadChip, which interrogates the methylation status of 485,577 CpGs in the human genome. We compared our methylation data with those from the TCGA (colorectal adenocarcinomas) 27K and 450K data. A near perfect adenoma/normal cluster and cancer samples was observed, indicating the significance and importance of epigenetic instability in the progression to colorectal cancer. We compared our top 140 hyper- and hypo-methylated genes with the TCGA 27K and 450K methylation data (data not shown). Our normal samples clustered with the normal from TCGA 27K and 450K data (both White and African American patients) and our tumors also clustered with the tumors from TCGA 27K and 450K data (both White and African American patient). This is consistent with our global methylation clustering (). However, TCGA 450K data showed more detailed alteration of gene methylation due to higher CpG density.Citation47 In addition, there seems to be some specificity in our gene methylation alteration vs. the TCGA 27K and 450K in regard to clinic pathological parameters among and within the samples.
Four CpG sites (SAP130, RAD54L, PFDN5, and PTPN12) were selected for further validation because they showed the largest differences between cancer and adenoma; all of these sites were hypermethylated in cancer. Additionally, these four CpG sites were located within the promoter region of the genes and were statistically significant based on the FDR adjusted P-values. Based on the criteria set forth in “Bioinformatics Approach #2,” these markers were considered to be eligible for further validation of the array data (, green box). Validation of RAD54L indicated that only 1 out of 40 patients showed hypermethylation (data not shown), suggesting that our 2nd bioinformatics approach was more stringent than the 1st approach and potentially better suited to selecting genes for validation.
CIMP- and non-CIMP-panel genes
Since we identified discrepancies in the selection process of genes with aberrant methylation based on IHM27 array data, we compiled and categorized lists of genes that have been found to be hypermethylated in CRC.
Thirty-five genes are among those used to identify CIMP phenotypes in CRC and were defined here as CRC CIMP gene-panel (). In addition to this panel, we have identified several new hypermethylated genes in CRC based on our review of the literature and are defined here as non-CIMP gene-panel (). The criteria we used for this 2nd panel were based on multiple citations found in the literature, which identified tumor suppressor genes using quantitative and qualitative methylation techniques.
Table 1. CIMP-panel genes
Table 2. Non-CIMP panel genes
Thus, we identified 16 genes (APC, EVL, CD109, PTEN, TWIST1, DCC, PTPRD, SFRP1, ICAM5, RASSF1A, EYA4, 30ST2, LAMA1, KCNQ5, ADHEF1, and TFPI2) that consistently showed promoter hypermethylation in CRC (). In our review, of the 16 non-CIMP gene-panel, APC and RASSF1A were the most cited markers (the majority of the studies used MSP or qMSP for the analysis of methylation). A difference in methylation assays is highlighted in the Kim et al. study (APC, ), which found only 15% methylationCitation6 in CRC compared with the other five studies that showed 92.6%, 42%, 47%, 27%, and 24% methylation of APC gene in various forms of CRC.Citation48 In addition to having the most citations, studies about CRC methylation in RASSF1A and APC include a total of 9 different ethnic populations, and both genes combined a range of methylation percentages that was large (15–93%). 3OST2 was cited to be > 50% methylated in CRC among the three studies, and 2 out of 3 studies used more than 100 patient samples and three different ethnic groups (). EVL was also cited to be > 50% methylated in CRC among 4 studies cited, and 3 out of 4 studies evaluated White American cohorts. Importantly, EVL was shown to be > 65% methylated in CRC in African Americans, suggesting that it may play a role in the more aggressive forms of CRC associated with African Americans (). CD109 and ICAM5 have the fewest citations in our review; however, the roles of genes that regulate extracellular matrix (ECM) are a hot topic in the field as they affect tumors’ invasiveness potential. Future studies are likely to identify similar genes involved in CRC. Although PTEN was only found to display low methylation in CRC in two studies (6% and 19%), the cohort sample sizes were large (n = 154, 126, and 146), suggesting high power in the statistics. Loss of PTEN is well documented in other cancers such as prostate. TWIST1 gene has been studied in three independent ethnic groups and is one of the more recently identified genes that is silenced by promoter methylation in CRC. DCC is an excellent candidate to be included in future CIMP panels as is highly methylated in CRC (82.7%, 80%, 56%, and 44%) in several studies. SFRP was shown to be methylated in colon adenoma and was cited as displaying > 90% methylation in CRC in 3 independent studies.Citation49-Citation52 EYA4 is one of the most recently identified genes found by four independent assays (MSP, methylation array, mass spectrometry, and pyrosequencing) to be hypermethylated in CRC.
Table 3. Summary of literature review for nonCIMP-panes genes
Discussion
It is now widely recognized that, in addition to genetic mutations, epigenetic mechanisms are involved in virtually every step of cancer development and progression, including in CRC. Methylation array technology offers high-throughput approaches to generating data about the methylation status of CpG sites within the genome; however, discrepancies exist in the evaluation of such data and the process for choosing individual genes (with CpG islands) for further validation. Herein, we present data that highlight the usefulness and limitations of using Infinium Illumina HumanMethylation27 array data and the validation of data using pyrosequencing.
In the statistical approach to analyze microarray data, we generated a lower 95% confidence limit value that correlated better with the level of methylation for an individual CpG site. In order to find those genes with CpG sites that are hypermethylated between groups (e.g., Cancer vs. Adenoma) we first ordered the data by P < 0.002 and Cancer Lower 95% CI > 0.9; for hypomethylated genes we ordered the data by P < 0.002 and Cancer Upper 95% CI < 0.1 (1st Bioinformatics Approach). Using this approach, we found that the hypermethylated genes had very small differences between the adenoma and cancer samples’ lower 95% CI, suggesting that although the statistical significance was high, the difference in absolute methylation was relatively small.
Since we observed very small differences in the lower 95% CI between groups using the 1st bioinformatics approach, we revised this approach (2nd Bioinformatic Approach) to first order the data by genes that showed the largest methylation differences between lower 95% CI followed by confirmation of statistical significance (FDR adjusted P-values < 0.05). This 2nd bioinformatics approach, was found to be more suitable for the detection of credible methylation targets between groups. While we recognize the limitations of the array in determining markers for differential methylation between samples, it is still a robust tool that can be used efficiently to cluster samples based on their global methylome profile. We and othersCitation53 report a very clear resolution of samples based on their global methylation signature. However, the major challenge remains on how to dissect these profiles for individual markers with high diagnostic and prognostic values. This is directly linked to the method used for data analysis. It is worth noting that the only adenoma that clustered with the cancer samples was of a tubulovillous type. The villous structure within colon adenoma points generally to advanced adenoma, further strengthening the usefulness of microarray as an efficient classifier of clinical samples. The TCGA 27K methylation data also showed a similar gene methylation profile when compared with our data, with some differences. Qualitative differences in the intensity distributions were probably driven by the clinicopathological differences among and within the samples.
The complications in validating our array data led us to prepare this manuscript with the inclusion of a review of the current literature with regards to candidate tumor suppressor genes in CRC that have been silenced by promoter methylation. Here we present a list of the genes that are currently used to categorize CRC via the CIMP parameters. Furthermore, we highlight 16 additional genes (APC, EVL, CD109, PTEN, TWIST1, DCC, PTPRD, SFRP1, ICAM5, RASSF1A, EYA4, 30ST2, LAMA1, KCNQ5, ADHEF1, and TFPI2) that have been shown to be hypermethylated in CRC, but are not yet used to evaluate the CIMP phenotype. An ingenuity pathway analysis (IPA) revealed that Wnt/b-catenin signaling was one of the top canonical pathways. However, the functions of the majority of these genes are associated with several critical pathways important in the initiation and progression of CRC.Citation54 These include the RAS2MAPK, PI3K, TGF-b, P53 and DNA mismatch-repair pathways.
In conclusion, there is a need for a systematic approach to choosing which genes to validate following the generation of high-throughput data. Moreover, the methylome signature of CRC should be updated more frequently. Efforts in these regards will contribute to the identification of prognostic markers to be used in early diagnosis, treatment and the evaluation of CRC. Such efforts must take advantage of new technologies such as NGS, for which the results are more reliable and comprehensive than for array-generated data.
Materials and Methods
Illumina HumanMethylation27 array
An Illumina HumanMethylation27 was used in this study. This array contains 27,578 CpG sites located at 14,495 genes promoter regions. DNA extracts from CRC, colon adenomas and normal tissues were modified using sodium bisulfite, fragmented and used in a microarray experiment using Illumina HumanMethylation27 array. All samples were collected after the Howard University Institutional Review Board’s approval of the study. DNA sample from each patient was fragmented and labeled with Cy3 or Cy5. The labeled DNA was applied to the array and hybridized for 40 h. The slides were washed and scanned. Prior to data analysis, the scanned slides underwent a Features Extraction process to generate the raw data for subsequent analysis
Bioinformatics
HumanMethylation27 DNA Analysis BeadChip (Illumina) was used to analyze CpG island methylation profiles in colorectal cancer (CRC) and adenoma. CpG island hypermethylation and hypomethylation in CRC was determined by the lower and upper 95% confidence interval values (β), respectively. A β value of 0–1 indicates the percentage of methylation from 0 to 100%.
Meta-analysis
A meta-analysis of DNA methylation in CRC was performed based on a review of the literature. First we identified several laboratories that were leaders in the field of CRC and compiled an initial list of genes found to be methylated in CRC by those labs (). Next, we categorized the genes into two groups: (1) genes currently used to characterize CRC as CIMP positive (i.e., CIMP-panel genes []), and (2) multiple citations in a PubMed search using keywords ([gene name] + “methylation” + “colorectal”). This process resulted in the identification of 16 genes () to be reviewed here.
Pyrosequencing
To design PCR and sequencing primers for Pyromark® Q24 instrument (Qiagen cat# 9001514) we used the PyroMark® Assay Design SW 2.0 (Qiagen cat# 9019077). To design pyrosequencing assays that interrogated the exact CpG site that was analyzed by the HumanMethylation27 array, we copied the sequence (approx. 150 bp) from “TOPGENOMICSEQUENCE” column of Illumina “HumanMethylation27_ 270596_v.1.2.cvs” file (downloaded from Illumina website), then pasted it into the PyroMark® Assay Design SW 2.0. The exact CpG site interrogated by the array is defined by brackets ([ ]). The bracketed CpG site was included in all pyrosequencing primer designs and surrounding CpG sites were included when possible. A total of 42 cycles were used for all PCR reactions and annealing temperatures were approximately 3°C below the lowest primer’s melting temperature. To confirm the integrity of PCR products, the amplicons were separated by electrophoresis on 2% agarose gel. PCR products (10 µl) were used in duplicates for pyrosequencing. Preparation of pyrosequencing reactions was performed and data analyzed as recommended by Qiagen.
Additional material
Download Zip (691.5 KB)Acknowledgments
"This project has been funded in whole or in part with Federal funds (UL1TR000101 previously UL1RR031975) from the National Center for Advancing Translational Sciences (NCATS), National Institutes of Health, through the Clinical and Translational Science Awards Program (CTSA), a trademark of DHHS, part of the Roadmap Initiative, “Re-Engineering the Clinical Research Enterprise.”, by NCI grant U01 CA162147, and RCMI.
Submitted
04/16/13
Revised
06/04/13
Accepted
06/21/13
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: http://www.landesbioscience.com/journals/epigenetics/article/25497
References
- Curtin K, Slattery ML, Samowitz WS. CpG island methylation in colorectal cancer: past, present and future. Patholog Res Int; 2011:902674.
- Kaneda A, Yagi K. Two groups of DNA methylation markers to classify colorectal cancer into three epigenotypes. Cancer Sci 2011; 102:18 - 24; http://dx.doi.org/10.1111/j.1349-7006.2010.01712.x; PMID: 21159060
- Kamiyama H, Noda H, Takata O, Suzuki K, Kawamura Y, Konishi F. Promoter hypermethylation of tumor-related genes in peritoneal lavage and the prognosis of patients with colorectal cancer. J Surg Oncol 2009; 100:69 - 74; http://dx.doi.org/10.1002/jso.21291; PMID: 19384904
- Lee BB, Lee EJ, Jung EH, Chun HK, Chang DK, Song SY, et al. Aberrant methylation of APC, MGMT, RASSF2A, and Wif-1 genes in plasma as a biomarker for early detection of colorectal cancer. Clin Cancer Res 2009; 15:6185 - 91; http://dx.doi.org/10.1158/1078-0432.CCR-09-0111; PMID: 19773381
- Naghibalhossaini F, Hosseini HM, Mokarram P, Zamani M. High frequency of genes’ promoter methylation, but lack of BRAF V600E mutation among Iranian colorectal cancer patients. Pathol Oncol Res 2011; 17:819 - 25; http://dx.doi.org/10.1007/s12253-011-9388-5; PMID: 21455633
- Kim JC, Choi JS, Roh SA, Cho DH, Kim TW, Kim YS. Promoter methylation of specific genes is associated with the phenotype and progression of colorectal adenocarcinomas. Ann Surg Oncol 2010; 17:1767 - 76; http://dx.doi.org/10.1245/s10434-009-0901-y; PMID: 20077021
- Fu X, Li J, Li K, Tian X, Zhang Y. Hypermethylation of APC promoter 1A is associated with moderate activation of Wnt signalling pathway in a subset of colorectal serrated adenomas. Histopathology 2009; 55:554 - 63; http://dx.doi.org/10.1111/j.1365-2559.2009.03411.x; PMID: 19912361
- Nagasaka T, Koi M, Kloor M, Gebert J, Vilkin A, Nishida N, et al. Mutations in both KRAS and BRAF may contribute to the methylator phenotype in colon cancer. Gastroenterology 2008; 134:1950 - 60, e1; http://dx.doi.org/10.1053/j.gastro.2008.02.094; PMID: 18435933
- Tokuyama Y, Takahashi T, Okumura N, Nonaka K, Kawaguchi Y, Yamaguchi K, et al. Aberrant methylation of heparan sulfate glucosamine 3-O-sulfotransferase 2 genes as a biomarker in colorectal cancer. Anticancer Res 2010; 30:4811 - 8; PMID: 21187457
- Gonzalo V, Lozano JJ, Muñoz J, Balaguer F, Pellisé M, Rodríguez de Miguel C, et al, Gastrointestinal Oncology Group of the Spanish Gastroenterological Association. Aberrant gene promoter methylation associated with sporadic multiple colorectal cancer. PLoS One 2010; 5:e8777; http://dx.doi.org/10.1371/journal.pone.0008777; PMID: 20098741
- Yi JM, Dhir M, Van Neste L, Downing SR, Jeschke J, Glöckner SC, et al. Genomic and epigenomic integration identifies a prognostic signature in colon cancer. Clin Cancer Res 2011; 17:1535 - 45; http://dx.doi.org/10.1158/1078-0432.CCR-10-2509; PMID: 21278247
- Chan TA, Glockner S, Yi JM, Chen W, Van Neste L, Cope L, et al. Convergence of mutation and epigenetic alterations identifies common genes in cancer that predict for poor prognosis. PLoS Med 2008; 5:e114; http://dx.doi.org/10.1371/journal.pmed.0050114; PMID: 18507500
- Mokarram P, Kumar K, Brim H, Naghibalhossaini F, Saberi-firoozi M, Nouraie M, et al. Distinct high-profile methylated genes in colorectal cancer. PLoS One 2009; 4:e7012; http://dx.doi.org/10.1371/journal.pone.0007012; PMID: 19750230
- Grady WM, Parkin RK, Mitchell PS, Lee JH, Kim YH, Tsuchiya KD, et al. Epigenetic silencing of the intronic microRNA hsa-miR-342 and its host gene EVL in colorectal cancer. Oncogene 2008; 27:3880 - 8; http://dx.doi.org/10.1038/onc.2008.10; PMID: 18264139
- Goel A, Arnold CN, Niedzwiecki D, Carethers JM, Dowell JM, Wasserman L, et al. Frequent inactivation of PTEN by promoter hypermethylation in microsatellite instability-high sporadic colorectal cancers. Cancer Res 2004; 64:3014 - 21; http://dx.doi.org/10.1158/0008-5472.CAN-2401-2; PMID: 15126336
- Arnold CN, Goel A, Blum HE, Boland CR. Molecular pathogenesis of colorectal cancer: implications for molecular diagnosis. Cancer 2005; 104:2035 - 47; http://dx.doi.org/10.1002/cncr.21462; PMID: 16206296
- Ahlquist T, Lind GE, Costa VL, Meling GI, Vatn M, Hoff GS, et al. Gene methylation profiles of normal mucosa, and benign and malignant colorectal tumors identify early onset markers. Mol Cancer 2008; 7:94; http://dx.doi.org/10.1186/1476-4598-7-94; PMID: 19117505
- Goel A, Nagasaka T, Arnold CN, Inoue T, Hamilton C, Niedzwiecki D, et al. The CpG island methylator phenotype and chromosomal instability are inversely correlated in sporadic colorectal cancer. Gastroenterology 2007; 132:127 - 38; http://dx.doi.org/10.1053/j.gastro.2006.09.018; PMID: 17087942
- Xu XL, Yu J, Zhang HY, Sun MH, Gu J, Du X, et al. Methylation profile of the promoter CpG islands of 31 genes that may contribute to colorectal carcinogenesis. World J Gastroenterol 2004; 10:3441 - 54; PMID: 15526363
- Ruppenthal RD, Nicolini C, Filho AF, Meurer R, Damin AP, Rohe A, et al. TWIST1 promoter methylation in primary colorectal carcinoma. Pathol Oncol Res 2011; 17:867 - 72; http://dx.doi.org/10.1007/s12253-011-9395-6; PMID: 21461979
- Oster B, Thorsen K, Lamy P, Wojdacz TK, Hansen LL, Birkenkamp-Demtröder K, et al. Identification and validation of highly frequent CpG island hypermethylation in colorectal adenomas and carcinomas. Int J Cancer 2011; 129:2855 - 66; http://dx.doi.org/10.1002/ijc.25951; PMID: 21400501
- Okada T, Suehiro Y, Ueno K, Mitomori S, Kaneko S, Nishioka M, et al. TWIST1 hypermethylation is observed frequently in colorectal tumors and its overexpression is associated with unfavorable outcomes in patients with colorectal cancer. Genes Chromosomes Cancer 2010; 49:452 - 62; PMID: 20140954
- Derks S, Bosch LJ, Niessen HE, Moerkerk PT, van den Bosch SM, Carvalho B, et al. Promoter CpG island hypermethylation- and H3K9me3 and H3K27me3-mediated epigenetic silencing targets the deleted in colon cancer (DCC) gene in colorectal carcinogenesis without affecting neighboring genes on chromosomal region 18q21. Carcinogenesis 2009; 30:1041 - 8; http://dx.doi.org/10.1093/carcin/bgp073; PMID: 19329758
- Ghoshal K, Motiwala T, Claus R, Yan P, Kutay H, Datta J, et al. HOXB13, a target of DNMT3B, is methylated at an upstream CpG island, and functions as a tumor suppressor in primary colorectal tumors. PLoS One 2010; 5:e10338; http://dx.doi.org/10.1371/journal.pone.0010338; PMID: 20454457
- Hibi K, Mizukami H, Shirahata A, Goto T, Sakata M, Sanada Y. Aberrant methylation of the netrin-1 receptor genes UNC5C and DCC detected in advanced colorectal cancer. World J Surg 2009; 33:1053 - 7; http://dx.doi.org/10.1007/s00268-008-9909-x; PMID: 19242752
- Hibi K, Sakata M, Sakuraba K, Kitamura YH, Shirahata A, Goto T, et al. Methylation of the DCC gene is lost in advanced gastric cancer. Anticancer Res 2010; 30:107 - 9; PMID: 20150623
- Shin SK, Nagasaka T, Jung BH, Matsubara N, Kim WH, Carethers JM, et al. Epigenetic and genetic alterations in Netrin-1 receptors UNC5C and DCC in human colon cancer. Gastroenterology 2007; 133:1849 - 57; http://dx.doi.org/10.1053/j.gastro.2007.08.074; PMID: 18054557
- Li VS, Yuen ST, Chan TL, Yan HH, Law WL, Yeung BH, et al. Frequent inactivation of axon guidance molecule RGMA in human colon cancer through genetic and epigenetic mechanisms. Gastroenterology 2009; 137:176 - 87; http://dx.doi.org/10.1053/j.gastro.2009.03.005; PMID: 19303019
- Rawson JB, Manno M, Mrkonjic M, Daftary D, Dicks E, Buchanan DD, et al. Promoter methylation of Wnt antagonists DKK1 and SFRP1 is associated with opposing tumor subtypes in two large populations of colorectal cancer patients. Carcinogenesis 2011; 32:741 - 7; http://dx.doi.org/10.1093/carcin/bgr020; PMID: 21304055
- Ahn JB, Chung WB, Maeda O, Shin SJ, Kim HS, Chung HC, et al. DNA methylation predicts recurrence from resected stage III proximal colon cancer. Cancer 2011; 117:1847 - 54; http://dx.doi.org/10.1002/cncr.25737; PMID: 21509761
- Tanaka J, Watanabe T, Kanazawa T, Tada T, Kazama Y, Tanaka T, et al. Silencing of secreted frizzled-related protein genes in MSI colorectal carcinogenesis. Hepatogastroenterology 2008; 55:1265 - 8; PMID: 18795670
- Worthley DL, Whitehall VL, Le Leu RK, Irahara N, Buttenshaw RL, Mallitt KA, et al. DNA methylation in the rectal mucosa is associated with crypt proliferation and fecal short-chain fatty acids. Dig Dis Sci 2011; 56:387 - 96; http://dx.doi.org/10.1007/s10620-010-1312-4; PMID: 20635146
- Suzuki H, Gabrielson E, Chen W, Anbazhagan R, van Engeland M, Weijenberg MP, et al. A genomic screen for genes upregulated by demethylation and histone deacetylase inhibition in human colorectal cancer. Nat Genet 2002; 31:141 - 9; http://dx.doi.org/10.1038/ng892; PMID: 11992124
- Lee S, Hwang KS, Lee HJ, Kim JS, Kang GH. Aberrant CpG island hypermethylation of multiple genes in colorectal neoplasia. Lab Invest 2004; 84:884 - 93; http://dx.doi.org/10.1038/labinvest.3700108; PMID: 15122305
- van Engeland M, Roemen GM, Brink M, Pachen MM, Weijenberg MP, de Bruïne AP, et al. K-ras mutations and RASSF1A promoter methylation in colorectal cancer. Oncogene 2002; 21:3792 - 5; http://dx.doi.org/10.1038/sj.onc.1205466; PMID: 12032847
- Wagner KJ, Cooper WN, Grundy RG, Caldwell G, Jones C, Wadey RB, et al. Frequent RASSF1A tumour suppressor gene promoter methylation in Wilms’ tumour and colorectal cancer. Oncogene 2002; 21:7277 - 82; http://dx.doi.org/10.1038/sj.onc.1205922; PMID: 12370819
- An B, Kondo Y, Okamoto Y, Shinjo K, Kanemitsu Y, Komori K, et al. Characteristic methylation profile in CpG island methylator phenotype-negative distal colorectal cancers. Int J Cancer 2010; 127:2095 - 105; http://dx.doi.org/10.1002/ijc.25225; PMID: 20131317
- Tommasi S, Pinto R, Petriella D, Pilato B, Lacalamita R, Santini D, et al. Oncosuppressor methylation: a possible key role in colon metastatic progression. J Cell Physiol 2011; 226:1934 - 9; http://dx.doi.org/10.1002/jcp.22524; PMID: 21506124
- Wahab AH, El-Mezayen HA, Sharad H, Rahman SA. Promoter hypermethylation of RASSF1A, MGMT, and HIC-1 genes in benign and malignant colorectal tumors. Tumour Biol 2011; http://dx.doi.org/10.1007/s13277-011-0156-7
- Osborn NK, Zou H, Molina JR, Lesche R, Lewin J, Lofton-Day C, et al. Aberrant methylation of the eyes absent 4 gene in ulcerative colitis-associated dysplasia. Clin Gastroenterol Hepatol 2006; 4:212 - 8; http://dx.doi.org/10.1016/j.cgh.2005.11.009; PMID: 16469682
- Schatz P, Distler J, Berlin K, Schuster M. Novel method for high throughput DNA methylation marker evaluation using PNA-probe library hybridization and MALDI-TOF detection. Nucleic Acids Res 2006; 34:e59; http://dx.doi.org/10.1093/nar/gkl218; PMID: 16670426
- Kim YH, Lee HC, Kim SY, Yeom YI, Ryu KJ, Min BH, et al. Epigenomic analysis of aberrantly methylated genes in colorectal cancer identifies genes commonly affected by epigenetic alterations. Ann Surg Oncol 2011; 18:2338 - 47; http://dx.doi.org/10.1245/s10434-011-1573-y; PMID: 21298349
- Varley KE, Mitra RD. Bisulfite Patch PCR enables multiplexed sequencing of promoter methylation across cancer samples. Genome Res 2010; 20:1279 - 87; http://dx.doi.org/10.1101/gr.101212.109; PMID: 20627893
- Kibriya MG, Raza M, Jasmine F, Roy S, Paul-Brutus R, Rahaman R, et al. A genome-wide DNA methylation study in colorectal carcinoma. BMC Med Genomics 2011; 4:50; http://dx.doi.org/10.1186/1755-8794-4-50; PMID: 21699707
- Glöckner SC, Dhir M, Yi JM, McGarvey KE, Van Neste L, Louwagie J, et al. Methylation of TFPI2 in stool DNA: a potential novel biomarker for the detection of colorectal cancer. Cancer Res 2009; 69:4691 - 9; http://dx.doi.org/10.1158/0008-5472.CAN-08-0142; PMID: 19435926
- Schnekenburger M, Diederich M. Epigenetics Offer New Horizons for Colorectal Cancer Prevention. Curr Colorectal Cancer Rep 2012; 8:66 - 81; http://dx.doi.org/10.1007/s11888-011-0116-z; PMID: 22389639
- Bibikova M, Barnes B, Tsan C, Ho V, Klotzle B, Le JM, et al. High density DNA methylation array with single CpG site resolution. Genomics 2011; 98:288 - 95; http://dx.doi.org/10.1016/j.ygeno.2011.07.007; PMID: 21839163
- Kim MS, Lee J, Sidransky D. DNA methylation markers in colorectal cancer. Cancer Metastasis Rev 2010; 29:181 - 206; http://dx.doi.org/10.1007/s10555-010-9207-6; PMID: 20135198
- Zhang W, Bauer M, Croner RS, Pelz JO, Lodygin D, Hermeking H, et al. DNA stool test for colorectal cancer: hypermethylation of the secreted frizzled-related protein-1 gene. Dis Colon Rectum 2007; 50:1618 - 26, discussion 1626-7; http://dx.doi.org/10.1007/s10350-007-0286-6; PMID: 17762966
- Lind GE, Danielsen SA, Ahlquist T, Merok MA, Andresen K, Skotheim RI, et al. Identification of an epigenetic biomarker panel with high sensitivity and specificity for colorectal cancer and adenomas. Mol Cancer 2011; 10:85; http://dx.doi.org/10.1186/1476-4598-10-85; PMID: 21777459
- Luo YX, Chen DK, Song SX, Wang L, Wang JP. Aberrant methylation of genes in stool samples as diagnostic biomarkers for colorectal cancer or adenomas: a meta-analysis. Int J Clin Pract 2011; 65:1313 - 20; http://dx.doi.org/10.1111/j.1742-1241.2011.02800.x; PMID: 22093539
- Simmer F, Brinkman AB, Assenov Y, Matarese F, Kaan A, Sabatino L, et al. Comparative genome-wide DNA methylation analysis of colorectal tumor and matched normal tissues. Epigenetics 2012; 7:1355 - 67; http://dx.doi.org/10.4161/epi.22562; PMID: 23079744
- Hinoue T, Weisenberger DJ, Lange CP, Shen H, Byun HM, Van Den Berg D, et al. Genome-scale analysis of aberrant DNA methylation in colorectal cancer. Genome Res 2012; 22:271 - 82; http://dx.doi.org/10.1101/gr.117523.110; PMID: 21659424
- Fearon ER. Molecular genetics of colorectal cancer. Annu Rev Pathol 2011; 6:479 - 507; http://dx.doi.org/10.1146/annurev-pathol-011110-130235; PMID: 21090969