Abstract
The oncogenic human papilloma viruses (HPVs) are associated with precancerous cervical lesions and development of cervical cancer. The DNA methylation signatures of the host genome in normal, precancerous and cervical cancer tissue may indicate tissue-specific perturbation in carcinogenesis. The aim of this study was to identify new candidate genes that are differentially methylated in squamous cell carcinoma compared with DNA samples from cervical intraepithelial neoplasia grade 3 (CIN3) and normal cervical scrapes. The Illumina Infinium HumanMethylation450 BeadChip method identifies genome-wide DNA methylation changes in CpG islands, CpG shores and shelves. Our findings showed an extensive differential methylation signature in cervical cancer compared with the CIN3 or normal cervical tissues. The identified candidate biomarker genes for cervical cancer represent several types of mechanisms in the cellular machinery that are epigenetically deregulated by hypermethylation, such as membrane receptors, intracellular signaling and gene transcription. The results also confirm extensive hypomethylation of genes in the immune system in cancer tissues. These insights into the functional role of DNA methylome alterations in cervical cancer could be clinically applicable in diagnostics and prognostics, and may guide the development of new epigenetic therapies.
Introduction
Cervical cancer is the third most frequent cancer among women world-wide.Citation1 The causative agent is a persistent infection with one of 15 oncogenic human papilloma viruses (HPVs) that leads to cervical precancerous lesions which may progress to cancer.Citation2 There are more than 100 identified HPV genotypes, 40 of them are considered to infect the genital tract and the types 16, 18, and 45 are found to be the most common in cervical cancer.Citation3
The majority of the viral load is cleared by the cell-mediated immunity, but if not cleared the persistent infection affects the host cell apoptosis and cell cycle control, cell adhesion and DNA repair mechanisms.Citation4 It may also activate inflammatory pathways, which was suggested to be critical for tumor development.Citation5
The integration of HPV virus in the host genome often occurs in the transcribed genomic region and this was suggested as a mechanism adapted by the virus to improve the expression of some viral products, notably E6 and E7 viral oncogenes.Citation6,Citation7 In addition, the integration of HPV type 16 was strongly associated with progression of precancerous lesions, i.e., cervical intraepithelial neoplasia grade 1 to 3 (CIN1‒CIN3).Citation8
Genomic instability and other DNA alterations including epigenetic ones, causing changed gene expression, are common features in many cancers.Citation9 Regions differentially methylated in cancers often co-localize with tissue-specific DNA methylated regions (T-DMR).Citation10,Citation11 In cervical cancer, DNA methylation can occur on the integrated viral DNA but aberrant DNA methylation is also induced in the host cell genome.Citation12 Increased methylation of HPV type 16 has been correlated with a more severe cancer progression.Citation13 Aberrant methylation status of several host genes related to cell cycle, apoptosis, development, cell adhesion and cellular signaling have been analyzed in clinical studies by Szalmaz et al. and Wentzen et al.Citation14,Citation15 These meta-studies also showed that there is a large variation in DNA methylation frequency, implying that new biomarkers are needed for early detection and progression of cervical cancer.
In this study, we employed the Illumina Infinium HumanMethylation450 BeadChip array to identify novel biomarker genes that are differentially methylated in cervical squamous cell carcinoma (SCC) DNA samples compared with DNA from normal cervical scrapes and CIN3 tissue. This approach enabled us to acquire a comprehensive epigenetic signature between the different cervical tissues in terms of global methylation, regional methylation in CpG islands (CGI), shores, gene related regions (TSS, 5′UTR, gene-body, 3′UTR), and also methylation of distinct CpG sites that discriminates between these tissues.
Results
DNA methylation profile in cervical cancer or CIN3 compared with normal cervical tissue
The probe call rate was > 99% for all of the samples and 454 215 CpG sites out of 485 577 were included in the analysis. The principal component analysis (PCA) analysis showed that all samples were close to each other. The majority of the cancer samples tended to hold together, followed by the cluster of CIN3 samples and the other cluster of the normal cervical samples (Fig. S1). The frequency distribution of the ∆β-values showed that cervical cancer tissue, compared with CIN3 and normal cervical tissue harbored hypermethylation at specific regions (data not shown). The frequency distribution of differentially methylated CpG sites across chromosomes showed that cancer tissue compared with the CIN3 and normal tissues had most differentially methylated CpG sites on chromosomes 1, 6, and 19 (Fig. S2).
Unsupervised hierarchical clustering analysis distinguished normal cervical tissues from tissues with CIN3 diagnosis and cancer tissues (). In this heatmap, 16 of the 25 statistically significant CpG sites belong to an annotated gene. The first ten CpG sites, located in different genes, are mostly hypomethylated in the cancer tissues as well as in the tissues with CIN3 diagnosis compared with the normal tissues (). In contrast, the remaining 15 CpG sites in specific genes are mostly hypermethylated both in cancer and tissues with CIN3 diagnosis compared with the normal tissues. The mean β-values for all the 25 CpG sites are shown in .
Figure 1. Unsupervised clustering analysis of the normal cervical tissue (N), CIN3 tissue (CIN3), and the cervical cancer tissue (CC). The color gradient green to red displays the β-value and can range from 0–1.
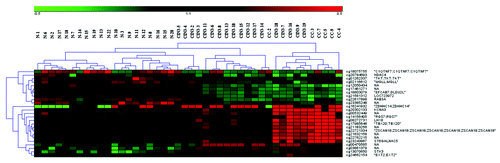
Table 1. Mean β-values for CpG sites represented in the heat map in
Differentially methylated CpG loci and gene regions
We analyzed the location of the hypo- and hyper-methylated CpG loci in relation to the CpG islands and gene context. Most of the differential methylation occurred in the context of a gene when comparing cancer tissue to precancerous and normal tissue (). A large fraction of the hypermethylated CpG loci were located in the CGI in contrast to the hypomethylated ones that were mostly located in the CGI shore or further away ().
Figure 2. Distribution of hypermethylated and hypomethylated CpG locus in CpG island (CGI), CpG island shore, CpG island shelves and other regions.
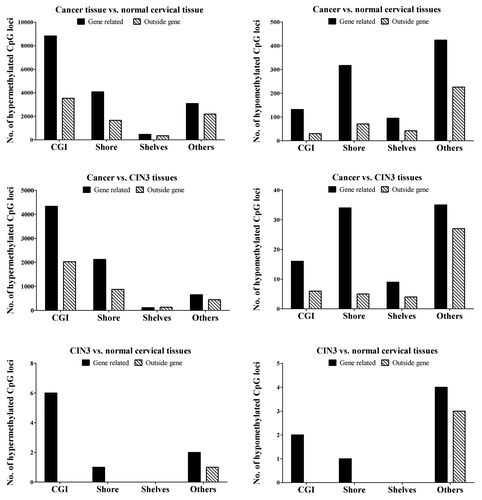
In Table S1 an overview of the number of differentially methylated regions and the range of the differences are shown comparing the three diagnostic groups. Most of the differentially methylated regions are found when comparing cancer tissue and normal cervical tissue. The majority of the events occur in the traditional gene promoter region (TSS1500, TSS200, 5′UTR, and 1st exon) and in the CpG islands. The comparison between cancer tissue and CIN3 tissues follow the same pattern. There are no differentially methylated regions reaching the criterion of ∆β of ≥ |0.2| when comparing the CIN3 tissues and normal cervical tissues.
Analysis of the differentially methylated regions in the vicinity of the traditional promoter region (TSS1500, TSS200, 5′UTR, and 1st exon) in cancer tissues compared with the normal tissues showed that many of the hypomethylated genes in cancer were found to be involved in the immune system response (). The hypermethylated genes are mostly receptors and transcription factors involved in cell development ().
Table 2. Hypomethylated gene promoter regions in cancer tissues compared with the normal tissues
Table 3. Hypermethylated gene promoter regions in cancer tissues compared with the normal tissues
Identification of functionally related gene groups
A total number of 74 gene ontology term clusters were generated when analyzing the hypomethylated CpG sites in cancer compared with normal cervical tissues. The GO_BP cluster with the highest enrichment score of 6.42 was related to the immune system (Table S2). The GO_FAT terms included several KEGG pathways involving the immune system, such as antigen processing and presentation, autoimmune thyroid disease and asthma.
The gene ontology analysis of the hypermethylated CpG sites created three clusters, and GO_BP had the highest enrichment score of 36.52 (Table S3). These gene ontology terms belong to the categories of cell development and cell differentiation. The GO_FAT and KEGG classification included categories of transcriptional regulation and DNA binding with the highest enrichment score of 24.41.
Candidate biomarker genes
The selection of candidate biomarker genes was based on both differentially methylated CpG sites generated with unsupervised hierarchical clustering and differentially methylated gene regions.
From the list of genes generated with unsupervised hierarchical clustering () the selection criteria were 1) Δβ-value > 0.2 between the cancer and normal tissues, and 2) a mean baseline (normal tissue) β-value < 0.2 of a CpG site hypermethylated in cancer or a baseline (normal tissue) β-value > 0.4 for a CpG site hypomethylated in cancer. This approach led to the selection of six genes, the best candidate biomarkers: RGS7, LHX8, STGALNAC5, TBX20, KCNA3, and ZSCAN18 ( and ), all with ≥ 9 significant CpG sites.
Table 4. Methylation status and gene expression of candidate biomarker genes
From and , displaying differentially methylated gene regions, we selected genes that were differentially methylated with a Δβ-value > |0.4|. The gene regions were then sorted according to the adjusted p-value and the top 20 regions were selected as candidates corresponding to 18 genes: ACAN, AJAP1, BARHL2, BOLL, C1orf114, FBXL7, GALR1, GYPC, KIF19, MIR663, PTGDR, S1PR4, SORCS1, TRIM58, TTYH1, VSTM2B, ZIK1, and ZNF582. The enlarged list of candidate biomarker genes is presented in Table 4. With exception of S1PR4, the candidate genes were hypermethylated in cancer compared with CIN3 and normal cervical tissues. As illustrative examples, the mean methylated fractions of all interrogated CpG sites in three selected genes, S1PR4, TBX20, and ZNF582 are shown in Figure S3. There is a stepwise increment in methylation (β-value) when comparing normal tissue, CIN3 tissues, and cervical cancer. We further validated the differential methylation of four selected genes, TBX20, RGS7, KCNA3, and S1PR4, with the Pyrosequencing assay technology. Figure S4 shows the percentage methylation of the analyzed CpG sites in cervical cancer and normal tissues. The results are in very good agreement with our array data. The mean methylation of the cervical cancer tissues was higher compared with the normal cervical tissues in the TBX20, RGS7, and KCNA3 genes, while the S1PR4 gene was hypomethylated in cervical cancer tissues. We found a higher variation in the methylated fraction within the cervical cancer group compared with the normal cervical tissues, as detected also by the array technique ().
External validation
External validation of our candidate biomarker genes was done using the mRNA expression data sets from Peyon et al.,Citation16 Scotto et al.,Citation17 and Zhai et al.,Citation18 which fulfilled quality criteria of tissue samples and employed high quality whole-genome expression arrays. Peyon et al.Citation16 reported probe sets for 21 of our 24 genes of interest, whereas the two other, reported sets for 14 of the genes. There was a good agreement between our DNA methylation data and the gene expression array: fold changes ranged between −1.09 and −2.20 for all hypermethylated genes, see . The hypomethylated S1PR4 gene was, as expected, upregulated in cervical cancer in one gene expression data set, but it was downregulated in the two other.
Discussion
The aim of this study was to acquire a comprehensive epigenetic signature of cervical cancer, and to identify new potential biomarker genes that are differentially methylated in cervical SCC compared with CIN3 or the normal cervical tissue. Our results showed that different cellular pathways are hypo- and hyper-methylated in cancer tissue. A short list of 24 novel candidate biomarker genes emerged, which was consistent with the external validation sets and could ultimately be clinically applicable.
Fifteen of the 24 potential biomarker genes (ACAN, Clorf114, FBXL7, GYPC, KCNA3, KIF19, LHX8, MIR663, RGS7, S1PR4, SORCS1, TBX20, TRIM58, TTYH1, and VSTM2B) have not yet been correlated with any cancer type, while eight (AJAP1, BARHL2, BOLL, GALR1, PTGDR, ST6GALNAC5, ZIK1, and ZSCAN18) have been implicated in cancers other than cervical cancer (). Nevertheless, all of these genes are included in crucial cellular functions, such as cell signaling, gene transcription, immunity, glycosylation, membrane activity and ubiquitination. Therefore, the hypermethylation of genes involved in such processes may contribute to the accumulation of damages that can progress to cervical cancer. By external validation using three independent data sets, the gene expression of 23 potential biomarkers was downregulated. This correlated with their hypermethylated state, confirming a link between DNA methylation and mRNA expression.
In cervical carcinogenesis, both global DNA hypomethylation and hypermethylation of specific genes promoters has been suggested to occur.Citation19,Citation20 We found here that most of the analyzed CpG loci were equally methylated (94%) in cervical cancer cells and normal cervical cells (Table S4). However, the genome-wide DNA methylation array confirmed that hypermethylation in cervical cancer occurs in fact at specific genes (24 199 CpG sites; Table S4). Even though we included more CIN3 cases precisely to increase the power to detect differences between CIN3 and normal tissues, the differences between the methylome of cervical cancer and normal tissues was greatest in all analyses (p- and β-values) compared with the methylome of CIN3 vs. normal tissues; therefore, our report focuses on these differences.
Distinctly different cellular pathways appear to be hypo- or hyper-methylated. In cervical cancer, the hypomethylated genes were related to the immune system response (Table S2), whereas the hypermethylated genes were involved in the biological processes of cell development, cell differentiation, and transcriptional regulation (Table S3). Shen et al.Citation21 interrogated a smaller number of CpG sites (Infinium Human Methylation27 BeadChip) in colon, kidney, stomach, lung and breast cancers as well as the corresponding normal tissue; they reported results consistent with our methylation data performed on cervical cancer.Citation21
Aberrant DNA methylation signatures in cervical cancer tissue, compared with the normal cervical tissue, are located in different regions depending on the methylation status. Hypermethylated CpG sites are predominantly located in the CGIs and hypomethylated CpG sites in the CGI shores (). In the gene context, most of the changes occurred in the TSS200 region (Table S1). Aberrant DNA methylation has been mostly studied in promoters with a CpG island. A few years back, CpG island shores were identified as potentially important regions harboring DNA methylation changes in colorectal cancer.Citation11 CpG island shore methylation was statistically significantly correlated with clinical characteristics in breast cancer when we analyzed the SLC25A43 gene.Citation22 Feber et al.Citation23 analyzed pooled DNA from benign and malignant nerve sheath tumors with methylated DNA immunoprecipitation and next generation sequencing (MeDIP-seq), and suggested that both hypo- and hyper-methylated differentially methylated regions were located in the CGI shore regions.Citation23 The MeDIP-seq methodology yields a semi-quantitative methylation percentage and differs from the Illumina Infinium HumanMethylation450 BeadChip that detects selected CpG sites at a single-nucleotide resolution in virtually all genes.Citation24,Citation25
Furthermore, methylation changes that are tumor driving were suggested to be present in premalignant lesions and to occur early in the process of carcinogenesis.Citation26 From a clinical perspective, methylation biomarkers for cervical cancer detection and prognosis are highly needed.Citation15 Lendvai et al.Citation19 recently used MeDIP-seq to perform a genome-wide analysis of high-grade CIN3 lesions compared with normal cervical epithelium, and identified 80 differentially methylated regions. They suggested COL25A1 and KATNAL2 genes as potential biomarkers for early detection of high-grade cervical intraepithelial neoplasia.Citation19 Our study also identified these two genes as differentially methylated in cervical cancer tissue compared with CIN3 or normal cervical tissue, but not when comparing CIN3 tissues and the normal cervical tissues (data not shown), indicating that these two genes might be late biomarkers, however they did not qualify as biomarkers according to our selection criteria. Our data extends the study of Lendivai et al.Citation19 by comparing the methylome of cervical cancer tissues compared with the CIN3 lesions and the normal tissues. By this approach we generated a list of 24 novel potential biomarker genes ().
One of our candidate biomarker gene, ZNF582, was previously shown to be hypermethylated in cervical cancer suggesting silencing of the ZNF582 gene in cervical neoplasms (Fig. S3).Citation27 Although the biological function of ZNF582 is not yet well characterized, it could be a tumor suppressor gene as well as potential candidate gene for molecular cervical cancer screening.Citation27 Another highly interesting gene is GALR1, which inhibits a key regulatory enzyme adenylyl cyclase. This gene was suggested to be hypermethylated in a subset of head and neck cancer caused by oncogenic HPV types, notably HPV 16.Citation28 Thus, frequent promoter hypermethylation of GALR1 and cellular growth suppression after re-expression of the gene supports the hypothesis that GALR1 could be a tumor suppressor gene in head and neck cancers,Citation28 as well as in cervical cancer (our study).
Our findings of hypomethylated genes in the immune system provide strong evidence that the immune response is under control of hypermethylation in normal cells, while in cancer cells, these genes become active by losing the methylation mark in the gene promoter region, which represses gene expression. One example is the hypomethylated AIM2 gene, whose functional protein forms an inflammasome, resulting in caspase activation in inflammatory cells. This induction of inflammatory cell response could occur in cervical cancer cells due to the hypomethylation of genes such as the AIM2 gene. Furthermore, AIM2 protein has been found overexpressed in a wide variety of tumor types, including neuroectodermal tumors,Citation29 pancreatic cancer,Citation30 and leukemias.Citation31
Another interesting hypomethylated gene is HLA-DPB1, whose gene product belongs to the HLA class II molecules, which plays a central role in the immune system by presenting peptides derived from extracellular proteins. There is evidence that HLA-DP participates in the functional T-cell responses against HPV in cervical cancer.Citation32 The host immune response to HPV is generally at low-level because HPV is shielded in the basal epithelial cells from the circulating immune cells during initial stages of the infection. Furthermore, HPV employs several mechanisms to downregulate innate and cell-mediated immunity.Citation33 This is confined to the early stages of infection since it is known that functionally active immune responses are generated at later stages of the HPV infection.Citation34 Thus, our findings in cervical cancer are consistent with an immune response against HPV, which is activated through hypomethylation of specific genes included in the immune response and inflammatory reactions.
The immune system, upon activation by hypomethylation, can prevent tumor development by suppressing the viral infection, prevent an inflammatory environment leading to tumorigenesis, and identify tumor cells based on their antigen expression and eliminate them.Citation35 Inflammatory cells are suggested to affect the tumor microenvironment, which in turn can promote or repress tumor growth.Citation36 The theoretical model of aberrant DNA methylation induced by chronic inflammation involves cytokine production TNFα, IL-1β, and oxidative stress in epithelial cells.Citation36,Citation37 Thus, without adequate immune responses, transformation to cancer might be quicker. This is the case of the adenovirus 12, which has the capability to turn off the immune system genes, which may lead to tumor progression.Citation38 Tumor tissue may contain increased amounts of leukocytes of various types, which could partly account for the observed hypomethylation of immune-system-related genes. The recent introduction into clinical practice of liquid-based cervical cytology may offer opportunities to address this issue in future studies.
In order to determine if DNA methylation changes occur early or late in cervical carcinogenesis, genome-wide DNA methylation studies should extend to lower CIN grades, CIN1 and CIN2 as well as atypical squamous cells of undetermined significance (ASCUS). However, our study clearly indicates that DNA methylation profile of specific genes in samples with high-grade cervical lesions (CIN3) is located between samples with normal diagnosis and samples with invasive cervical cancer (Fig. S1), but closer to cluster of normal samples, as exemplified by specific CpG sites in the ZNF582, TBX20, and S1PR4 genes (Fig. S3).
We have validated our array results with an independent method analyzing the methylated fraction of four genes (Fig. S4). Our results suggest as previously reported by Cachill et al.Citation39 and Deneberg et al.,Citation40 that the Illumina HumanMethylation450 array measures differential DNA methylation comparable to the golden standard pyrosequencing assay technology.
Conclusion
In this study, we selected candidate genes with CpG sites highly methylated in cervical cancer tissue and without methylation in normal cervical tissue. This combination provides an optimal condition for a successful diagnostic test. The results highlight the extensive differential methylation signature in HPV-induced cervical cancer tissue compared with CIN3 or normal cervical tissues. The 24 candidate biomarkers that we identified represent several types of mechanisms in the cellular machinery that are epigenetically deregulated, such as membrane receptor function, intracellular signaling, and gene transcription. External validation confirmed a link between DNA methylation and mRNA levels for these genes. Considering the extensive hypomethylation of genes involved in the immune system, epigenetic therapy and immune therapy might need to be combined when treating cervical cancer. Understanding of the functional role of DNA methylation alterations in cancer genomes may prove to be clinically applicable in disease diagnostics and prognostics, and may guide the development of new epigenetic therapies.
Materials and Methods
Study group
The cervical specimens were collected in the Sisters of Mercy Hospital, Zagreb, Croatia during the period from 2004 to 2011. The clinical characteristics of the specimens are presented in Table S5. The cytological diagnoses were obtained for normal cervical smears, high-grade squamous intraepithelial lesion (HSIL)/CIN3, while cervical squamous cell carcinoma (SCC) samples were defined by histopathology.Citation41 Subsequently, cervical specimens were tested and typed for HPV.Citation42
The cervical sample collection is regulated through laboratory service request forms that have to be signed and approved by the practicing physician. The extracted DNA from cervical specimens was processed without initial knowledge of patient data (age, diagnosis, HPV detection, and typing result). The study was approved by the Ethical Board of the Rudjer Boskovic Institute, as well as the Ethical Board of Sisters of Mercy Hospital, and conforms to the Helsinki declaration (DoH/Oct2008). The Ethical Review Board, Uppsala, approved the Swedish participation in this study.
DNA preparation
DNA from cervical cell samples was isolated on the BioRobot EZ1 (Qiagen, Hilden, Germany) according to the manufacturer’s instruction. After DNA extraction, the purified DNA was dissolved in 50–100 μl of tri-distillate sterile water and stored at –20 °C until further analysis. Each DNA was analyzed by electrophoresis on 1% agarose gels and spectrophotometrically.Citation43
HPV detection and typing
Three sets of consensus primers for HPV detection were used: PGMY09/PGMY11,Citation44 L1C1/L1C2-1/L1C2-2Citation45 and GP5+/GP6+.Citation46 The quality of the isolated DNA was tested by amplification of the 268 bp sequence of the β-globin gene using PC04/GH20 primersCitation47 in a multiplex PCR with PGMY primers. Type-specific primers for HPV types 6/11, 16, 18, 31, 33, 45, 52, and 58 were used for HPV typing according to Milutin-Gasperov et al., 2007.Citation42 Aliquots of each PCR product (10 μl) were analyzed by electrophoresis on 2% agarose gels stained with ethidium bromide. The amplified products were visualized by UV irradiation of the gels either by ImageMaster VDS (Pharmacia Biotech) or UVItec Cambridge (Alliance 4.7).
Methylation array
The Illumina Infinium HumanMethylation450 BeadChip (Illumina, Sweden) includes 485,577 CpG sites located all over the genome.Citation24 The quality and integrity of the samples were evaluated with Nano Drop and gel electrophoresis; samples were included if the ratio A260/280 was between 1.7–1.9, the ratio A 260/230 was > 1 and the samples resolved on gel electrophoresis showed a distinct DNA band > 10 000 bp. In this study, 20 normal cervical samples (HPV negative), 18 samples with CIN3 lesions (HPV positive) and 6 cervical cancer tissues (HPV positive) were included. The DNA methylation assay was performed as follows. Briefly, approximately 500 ng of DNA from cancer, CIN3 lesions, and normal cervical tissues were bisulfite treated with Zymo EZ DNA Methylation Gold kit (Zymo Research) according to manufacturer’s procedure. After the bisulfite treatment, 200 ng DNA was subjected to the whole genome amplification (WGA) and enzymatic digestion with reagents provided within the Infinium HumanMethylation450 kit (Illumina). The hybridization of the samples on the BeadChips and washing procedures followed the standard procedures obtained from Illumina. The iScan scanner (Illumina) was used to read the BeadChips and the data collection was performed in the GenomeStudio software (version 1.0).
Data processing
The Infinium Human 450K Methylation Bead chip has several built in sample dependent and independent controls that measure the processing of the chip. They were analyzed in the Genome Studio software with the Dash-Board module, prior to further statistical analyzes. The probe call rate for a passed sample should be > 98%, and the detection p-value for the probes should be < 0.01, otherwise they were excluded. The β-value generated for each CpG locus measures the intensity of methylated (β = 1) and unmethylated probes (β = 0). It is calculated as β = [M/(M + U + 100)], where M = methylated allele and U = unmethylated allele. The study cohort contains only women; therefore the Y-chromosome was excluded from the analysis. The raw β-values were generated with the GenomeBead studio using the settings normalization controls and background subtraction. This data has been deposited to the Gene Expression Omnibus database under the accession number GSE46306.
A CpG locus was considered differentially methylated if the Δβ-value was ≥ ∣0.2∣ and the adjusted P value < 0.05. This cut-off value of ∣0.2∣ represents the 99% confidence interval of the detection limit.Citation25
Unsupervised clustering of CpG loci and DAVID analysis
Heat map showing clustering of the patient samples was performed based on P value < 0.05 and ∆β-value ± 0.5 between CIN3 tissues and normal tissues. They were produced with the Illumina Methylation Analyzer (IMA) package version 2.1.1 (Illumina). The Database for Annotation, Visualization and Integrated Discovery (DAVID)Citation48 was used to analyze biological features associated with genes hypo- or hypermethylated in cervical cancer compared with normal cervical tissue. The gene lists were obtained with the criteria P value < 0.05 and a more stringent Δβ-value of > |0.3| (between cancer and normal tissues) due to the input limit of genes to the DAVID tool. The gene ontology (GO) groups were clustered into functional annotation referring to biological process (GO_BP), cellular component (GO_CC) and molecular function (GO_MF). GO_FAT terms were also retrieved to filter the broadest GO categories. In addition, the Kyoto Encyclopedia of Genes and Genomes (KEGG) database was used to identify functional gene groups and ontology terms that are significantly overrepresented.
Differentially methylated CpG loci and gene regions
The CpG loci were categorized as either hypermethylated (positive Δβ-value) or hypomethylated (negative Δβ-value). The CpG loci were then categorized in the context of (I) CpG islands, CpG shores, CpG shelves or others and (II) genomic location of TSS1500 (1.5 kb within transcriptional start site), TSS200 (200 bp within transcriptional start site), 5′UTR (5′ untranslated region) and 1st exon, gene body and 3′UTR (3′ untranslated region) or others. To compare the regions (CpG island, CpG shore, CpG shelves, TSS1500, TSS200, 5′UTR, 1st exon, gene body, and 3′UTR) between the different diagnostic groups, the median β-value was calculated including all the CpG sites in the analyzed region. Each region contains on average 1.53–9.92 probes.Citation25
External validation of the candidate biomarker genes
Three data sets GSE6791,Citation16 GSE9750,Citation17 and GSE7803Citation18 deposited to the Gene Expression Omnibus (GEO) database were used for external validation of mRNA expression of candidate biomarker genes. The data sets measured gene expression by the Affymetrix GeneChip Human genome U133 Plus 2.0 (GSE6791) array and Affymetrix HG-U133A array (GSE9750 and GSE7803). The GSE6791data set was composed of 20 cervical cancer samples (17 HPV+, 3 HPV-) and 8 normal cervical samples (HPV-); the GSE9750 data set of 27 cervical cancers (HPV+) and 24 normal cervical tissues (HPV-); and GSE7803 data set of 24 cervical cancer tissues (HPV+) and 10 normal cervical tissues (HPV-). Using the online tool GEO2R (www.ncbi.nlm.nih.-gov/geo/geo2r), the log fold change (FC) was calculated to estimate the differences in the gene expression in cervical cancer compared with the normal cervical samples. The unadjusted P values < 0.05 were considered as statistically significant, since these analyses were hypothesis driven.
Pyrosequencing validation of differential methylation
We developed assays for the following genes: TBX20, RGS7, KCNA3, and S1PR4. The PCR and sequencing primers were designed to assess the exactly same CpG sites analyzed by the array. All primers were purchased from www.biomers.net (Ulm/Donau). In the Table S6, the sequences, amplicon sizes, and the optimal annealing temperatures are indicated.
The analysis was performed on 5 normal and 5 cancer cervical tissues, which were already tested by Illumina Infinium HumanMethylation450 BeadChip array. Briefly, approximately 500 ng extracted DNA was used for the bisulfite treatment performed with the EZ DNA Methylation Gold kit according to the instructions by the manufacturer and eluted in 20 µl elution buffer (Zymo Research, Orion Diagnostica). The PCR reactions were performed in a total volume of 40 µl with the KAPPA2G Robust HotStarTaq DNA Polymerase Kit (Kapa Biosystems, Inc.), containing 0.10 µmol/L of each primer, 0.5 units of Taq polymerase, 1.5 mM MgCl2, and 0.1 mM each of dGTP, dATP, dTTP, dCTP and 50 ng of bisulphite treated DNA was added as template. The PCR reaction for the RGS7 gene contained also the 1X Enhancer solution provided in the kit. The PCR program was as follows: initial denaturation step of 1 min at 95 °C, followed by 45 cycles of 30 s denaturation at 95 °C, specific annealing Ta for 30 s and extension for 30 s at 72 °C and one cycle of final extension for 30 s at 72 °C. Sequencing was performed using a Pyromark Gold Q96 Reagent Kit and a PSQ 96ID system (Qiagen) as described previously by Farkas et al.Citation49 The nucleotide addition order was optimized by the Pyro Q-CpG software version 1.0.9 (Qiagen) and the results were automatically analyzed using the same software.
Statistical analysis
All statistical analyses were performed using the R software with the IMA package.Citation50 The raw β-values were arcsine square root transformed and the empirical Bayes moderated t-statistic was used to generate the P values.Citation51 The Benjamini-Hochberg method was used to adjust the P values for multiple testing.Citation52 For site-level analysis the mean Δβ-values were used and for the regional analysis median Δβ-values value was calculated including all CpG sites in one region. The principal component analysis (PCA) was performed to visualize the difference in DNA methylation between the cancer and normal tissue samples. In the DAVID analysis the statistic used was the modified Fishers exact P value, adjusted for multiple testing with the Bonferroni method.Citation53
| Abbreviations: | ||
| CGI | = | CpG island |
| CIN3 | = | cervical intraepithelial neoplasia grade |
| HPV | = | human papilloma virus |
| PCA | = | principal component analysis |
| SCC | = | squamous cell carcinoma |
| T-DMR | = | tissue-specific differentially methylated regions |
Additional material
Download Zip (2 MB)Acknowledgments
Methylation profiling was performed by the SNP&SEQ Technology Platform in Uppsala. The platform is part of Science for Life Laboratory at Uppsala University and supported as a national infrastructure by the Swedish Research Council. This work was supported by Lions Cancer Foundation, Nyckelfonden, Örebro läns landsting (Farkas SA and Nilsson TK), and partially by the Croatian Ministry of Science, Education and Sports (Grant number: 098-0982464-2510; Grce, M and Milutin-Gašperov, N).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/epigenetics/article/26346
References
- Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin 2011; 61:69 - 90; http://dx.doi.org/10.3322/caac.20107; PMID: 21296855
- Schiffman M, Castle PE, Jeronimo J, Rodriguez AC, Wacholder S. Human papillomavirus and cervical cancer. Lancet 2007; 370:890 - 907; http://dx.doi.org/10.1016/S0140-6736(07)61416-0; PMID: 17826171
- Munoz N, Castellsague X, de Gonzalez AB, Gissmann L. HPV in the etiology of human cancer. Vaccine 2006; 24:1 - 10; http://dx.doi.org/10.1016/j.vaccine.2006.05.115; PMID: 16122853
- Whiteside MA, Siegel EM, Unger ER. Human papillomavirus and molecular considerations for cancer risk. Cancer 2008; 113:Suppl 2981 - 94; http://dx.doi.org/10.1002/cncr.23750; PMID: 18980282
- Coussens LM, Werb Z. Inflammation and cancer. Nature 2002; 420:860 - 7; http://dx.doi.org/10.1038/nature01322; PMID: 12490959
- Schmitz M, Driesch C, Jansen L, Runnebaum IB, Dürst M. Non-random integration of the HPV genome in cervical cancer. PLoS One 2012; 7:e39632; http://dx.doi.org/10.1371/journal.pone.0039632; PMID: 22761851
- Ziegert C, Wentzensen N, Vinokurova S, Kisseljov F, Einenkel J, Hoeckel M, von Knebel Doeberitz M. A comprehensive analysis of HPV integration loci in anogenital lesions combining transcript and genome-based amplification techniques. Oncogene 2003; 22:3977 - 84; http://dx.doi.org/10.1038/sj.onc.1206629; PMID: 12813471
- Li W, Wang W, Si M, Han LF, Gao QL, Luo AY, Li Y, Lu Y, Wang S, Ma D. The physical state of HPV16 infection and its clinical significance in cancer precursor lesion and cervical carcinoma. J Cancer Res Clin Oncol 2008; 134:1355 - 61; http://dx.doi.org/10.1007/s00432-008-0413-3; PMID: 18478264
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144:646 - 74; http://dx.doi.org/10.1016/j.cell.2011.02.013; PMID: 21376230
- Hansen KD, Timp W, Bravo HC, Sabunciyan S, Langmead B, McDonald OG, Wen B, Wu H, Liu Y, Diep D, et al. Increased methylation variation in epigenetic domains across cancer types. Nat Genet 2011; 43:768 - 75; http://dx.doi.org/10.1038/ng.865; PMID: 21706001
- Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, Cui H, Gabo K, Rongione M, Webster M, et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet 2009; 41:178 - 86; http://dx.doi.org/10.1038/ng.298; PMID: 19151715
- Yanatatsaneejit P, Mutirangura A, Kitkumthorn N. Human papillomavirus’s physical state and cyclin A1 promoter methylation in cervical cancer. Int J Gynecol Cancer 2011; 21:902 - 6; http://dx.doi.org/10.1097/IGC.0b013e3182158683; PMID: 21412159
- Mazumder Indra D, Singh RK, Mitra S, Dutta S, Chakraborty C, Basu PS, Mondal RK, Roychoudhury S, Panda CK. Genetic and epigenetic changes of HPV16 in cervical cancer differentially regulate E6/E7 expression and associate with disease progression. Gynecol Oncol 2011; 123:597 - 604; http://dx.doi.org/10.1016/j.ygyno.2011.08.004; PMID: 21911249
- Szalmás A, Kónya J. Epigenetic alterations in cervical carcinogenesis. Semin Cancer Biol 2009; 19:144 - 52; http://dx.doi.org/10.1016/j.semcancer.2009.02.011; PMID: 19429477
- Wentzensen N, Sherman ME, Schiffman M, Wang SS. Utility of methylation markers in cervical cancer early detection: appraisal of the state-of-the-science. Gynecol Oncol 2009; 112:293 - 9; http://dx.doi.org/10.1016/j.ygyno.2008.10.012; PMID: 19054549
- Pyeon D, Newton MA, Lambert PF, den Boon JA, Sengupta S, Marsit CJ, Woodworth CD, Connor JP, Haugen TH, Smith EM, et al. Fundamental differences in cell cycle deregulation in human papillomavirus-positive and human papillomavirus-negative head/neck and cervical cancers. Cancer Res 2007; 67:4605 - 19; http://dx.doi.org/10.1158/0008-5472.CAN-06-3619; PMID: 17510386
- Scotto L, Narayan G, Nandula SV, Arias-Pulido H, Subramaniyam S, Schneider A, Kaufmann AM, Wright JD, Pothuri B, Mansukhani M, et al. Identification of copy number gain and overexpressed genes on chromosome arm 20q by an integrative genomic approach in cervical cancer: potential role in progression. Genes Chromosomes Cancer 2008; 47:755 - 65; http://dx.doi.org/10.1002/gcc.20577; PMID: 18506748
- Zhai Y, Kuick R, Nan B, Ota I, Weiss SJ, Trimble CL, Fearon ER, Cho KR. Gene expression analysis of preinvasive and invasive cervical squamous cell carcinomas identifies HOXC10 as a key mediator of invasion. Cancer Res 2007; 67:10163 - 72; http://dx.doi.org/10.1158/0008-5472.CAN-07-2056; PMID: 17974957
- Lendvai A, Johannes F, Grimm C, Eijsink JJH, Wardenaar R, Volders HH, Klip HG, Hollema H, Jansen RC, Schuuring E, et al. Genome-wide methylation profiling identifies hypermethylated biomarkers in high-grade cervical intraepithelial neoplasia. Epigenetics 2012; 7:1268 - 78; http://dx.doi.org/10.4161/epi.22301; PMID: 23018867
- Kim YI, Giuliano A, Hatch KD, Schneider A, Nour MA, Dallal GE, Selhub J, Mason JB. Global DNA hypomethylation increases progressively in cervical dysplasia and carcinoma. Cancer 1994; 74:893 - 9; http://dx.doi.org/10.1002/1097-0142(19940801)74:3<893::AID-CNCR2820740316>3.0.CO;2-B; PMID: 8039116
- Shen XP, He Z, Li HD, Yao C, Zhang Y, He L, Li S, Huang J, Guo Z. Distinct functional patterns of gene promoter hypomethylation and hypermethylation in cancer genomes. PLoS One 2012; 7:e44822; http://dx.doi.org/10.1371/journal.pone.0044822; PMID: 22970311
- Lindqvist BM, Farkas SA, Wingren S, Nilsson TK. DNA methylation pattern of the SLC25A43 gene in breast cancer. Epigenetics 2012; 7:300 - 6; http://dx.doi.org/10.4161/epi.7.3.19064; PMID: 22430806
- Feber A, Wilson GA, Zhang L, Presneau N, Idowu B, Down TA, Rakyan VK, Noon LA, Lloyd AC, Stupka E, et al. Comparative methylome analysis of benign and malignant peripheral nerve sheath tumors. Genome Res 2011; 21:515 - 24; http://dx.doi.org/10.1101/gr.109678.110; PMID: 21324880
- Sandoval J, Heyn HA, Moran S, Serra-Musach J, Pujana MA, Bibikova M, Esteller M. Validation of a DNA methylation microarray for 450,000 CpG sites in the human genome. Epigenetics 2011; 6:692 - 702; http://dx.doi.org/10.4161/epi.6.6.16196; PMID: 21593595
- Bibikova M, Barnes B, Tsan C, Ho V, Klotzle B, Le JM, Delano D, Zhang L, Schroth GP, Gunderson KL, et al. High density DNA methylation array with single CpG site resolution. Genomics 2011; 98:288 - 95; http://dx.doi.org/10.1016/j.ygeno.2011.07.007; PMID: 21839163
- Kalari S, Pfeifer GP. Identification of driver and passenger DNA methylation in cancer by epigenomic analysis. Adv Genet 2010; 70:277 - 308; http://dx.doi.org/10.1016/B978-0-12-380866-0.60010-1; PMID: 20920752
- Huang RL, Chang CC, Su PH, Chen YC, Liao YP, Wang HC, Yo YT, Chao TK, Huang HC, Lin CY, et al. Methylomic analysis identifies frequent DNA methylation of zinc finger protein 582 (ZNF582) in cervical neoplasms. PLoS One 2012; 7:e41060; http://dx.doi.org/10.1371/journal.pone.0041060; PMID: 22815913
- Misawa K, Ueda Y, Kanazawa T, Misawa Y, Jang I, Brenner JC, Ogawa T, Takebayashi S, Grenman RA, Herman JG, et al. Epigenetic inactivation of galanin receptor 1 in head and neck cancer. Clin Cancer Res 2008; 14:7604 - 13; http://dx.doi.org/10.1158/1078-0432.CCR-07-4673; PMID: 19047085
- Ge L, Cornforth AN, Hoa NT, Delgado C, Chiou SK, Zhou YH, Jadus MR. Differential glioma-associated tumor antigen expression profiles of human glioma cells grown in hypoxia. PLoS One 2012; 7:e42661; http://dx.doi.org/10.1371/journal.pone.0042661; PMID: 22957023
- Pedersen KS, Bamlet WR, Oberg AL, de Andrade M, Matsumoto ME, Tang H, Thibodeau SN, Petersen GM, Wang L. Leukocyte DNA methylation signature differentiates pancreatic cancer patients from healthy controls. PLoS One 2011; 6:e18223; http://dx.doi.org/10.1371/journal.pone.0018223; PMID: 21455317
- Yamazaki J, Taby R, Vasanthakumar A, Macrae T, Ostler KR, Shen L, Kantarjian HM, Estecio MR, Jelinek J, Godley LA, et al. Effects of TET2 mutations on DNA methylation in chronic myelomonocytic leukemia. Epigenetics 2012; 7:201 - 7; http://dx.doi.org/10.4161/epi.7.2.19015; PMID: 22395470
- Piersma SJ, Welters MJ, van der Hulst JM, Kloth JN, Kwappenberg KM, Trimbos BJ, Melief CJ, Hellebrekers BW, Fleuren GJ, Kenter GG, et al. Human papilloma virus specific T cells infiltrating cervical cancer and draining lymph nodes show remarkably frequent use of HLA-DQ and -DP as a restriction element. Int J Cancer 2008; 122:486 - 94; http://dx.doi.org/10.1002/ijc.23162; PMID: 17955486
- Moscicki AB, Schiffman M, Kjaer S, Villa LL. Chapter 5: Updating the natural history of HPV and anogenital cancer. Vaccine 2006; 24 Suppl 3:S3/42-51.
- Feller L, Wood NH, Khammissa RA, Chikte UM, Meyerov R, Lemmer J. HPV modulation of host immune responses. SADJ 2010; 65:266 - 8; PMID: 20879650
- Swann JB, Smyth MJ. Immune surveillance of tumors. J Clin Invest 2007; 117:1137 - 46; http://dx.doi.org/10.1172/JCI31405; PMID: 17476343
- Lu HT, Ouyang WM, Huang CS. Inflammation, a key event in cancer development. Mol Cancer Res 2006; 4:221 - 33; http://dx.doi.org/10.1158/1541-7786.MCR-05-0261; PMID: 16603636
- Chiba T, Marusawa H, Ushijima T. Inflammation-associated cancer development in digestive organs: mechanisms and roles for genetic and epigenetic modulation. Gastroenterology 2012; 143:550 - 63; http://dx.doi.org/10.1053/j.gastro.2012.07.009; PMID: 22796521
- Doerfler W. Adenoviruses. In: Baron S, ed. Medical Microbiology. Galveston (TX): University of Texas Medical Branch at Galveston The University of Texas Medical Branch at Galveston, 1996.
- Cahill N, Bergh AC, Kanduri M, Göransson-Kultima H, Mansouri L, Isaksson A, Ryan F, Smedby KE, Juliusson G, Sundström C, et al. 450K-array analysis of chronic lymphocytic leukemia cells reveals global DNA methylation to be relatively stable over time and similar in resting and proliferative compartments. Leukemia 2013; 27:150 - 8; http://dx.doi.org/10.1038/leu.2012.245; PMID: 22922567
- Deneberg S, Guardiola P, Lennartsson A, Qu Y, Gaidzik V, Blanchet O, Karimi M, Bengtzén S, Nahi H, Uggla B, et al. Prognostic DNA methylation patterns in cytogenetically normal acute myeloid leukemia are predefined by stem cell chromatin marks. Blood 2011; 118:5573 - 82; http://dx.doi.org/10.1182/blood-2011-01-332353; PMID: 21960591
- Ovanin-Rakic A, Pajtler M, Stanković T, Audy-Jurković S, Ljubojević N, Grubišić G, et al. The classification of cytologic findings of cervix uteri “Zagreb 2002”: The Modification of the “Zagreb 1990” and “NCI Bethesda System 2001” Classifications. Gynaecologia Et Perinatologia 2003; 12:148 - 53
- Milutin-Gasperov N, Sabol I, Halec G, Matovina M, Grce M. Retrospective study of the prevalence of high-risk human papillomaviruses among Croatian women. Coll Antropol 2007; 31:Suppl 2 89 - 96; PMID: 17598510
- J S. EF F, T M. Molecular cloning - A laboratory manual. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory, 1982.
- Gravitt PE, Peyton CL, Alessi TQ, Wheeler CM, Coutlée F, Hildesheim A, Schiffman MH, Scott DR, Apple RJ. Improved amplification of genital human papillomaviruses. J Clin Microbiol 2000; 38:357 - 61; PMID: 10618116
- Yoshikawa H, Kawana T, Kitagawa K, Mizuno M, Yoshikura H, Iwamoto A. Detection and typing of multiple genital human papillomaviruses by DNA amplification with consensus primers. Jpn J Cancer Res 1991; 82:524 - 31; http://dx.doi.org/10.1111/j.1349-7006.1991.tb01882.x; PMID: 1648051
- van den Brule AJ, Pol R, Fransen-Daalmeijer N, Schouls LM, Meijer CJ, Snijders PJ. GP5+/6+ PCR followed by reverse line blot analysis enables rapid and high-throughput identification of human papillomavirus genotypes. J Clin Microbiol 2002; 40:779 - 87; http://dx.doi.org/10.1128/JCM.40.3.779-787.2002; PMID: 11880393
- Bell DA, Taylor JA, Paulson DF, Robertson CN, Mohler JL, Lucier GW. Genetic risk and carcinogen exposure: a common inherited defect of the carcinogen-metabolism gene glutathione S-transferase M1 (GSTM1) that increases susceptibility to bladder cancer. J Natl Cancer Inst 1993; 85:1159 - 64; http://dx.doi.org/10.1093/jnci/85.14.1159; PMID: 8320745
- Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009; 4:44 - 57; http://dx.doi.org/10.1038/nprot.2008.211; PMID: 19131956
- Farkas SA, Böttiger AK, Isaksson HS, Finnell RH, Ren A, Nilsson TK. Epigenetic alterations in folate transport genes in placental tissue from fetuses with neural tube defects and in leukocytes from subjects with hyperhomocysteinemia. Epigenetics 2013; 8:303 - 16; http://dx.doi.org/10.4161/epi.23988; PMID: 23417011
- Wang D, Yan L, Hu Q, Sucheston LE, Higgins MJ, Ambrosone CB, Johnson CS, Smiraglia DJ, Liu S. IMA: an R package for high-throughput analysis of Illumina’s 450K Infinium methylation data. Bioinformatics 2012; 28:729 - 30; http://dx.doi.org/10.1093/bioinformatics/bts013; PMID: 22253290
- Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 2004; 3:e3; http://dx.doi.org/10.2202/1544-6115.1027; PMID: 16646809
- Benjamini Y, Hochberg Y. Controlling the false discovery rate - a practical and poerful approach to multiple testing. J R Stat Soc, B 1995; 57:289 - 300
- Holm S. A simple sequentially rejective multiple test procedure. Scand J Stat 1979; 6:65 - 70
- Jeffries MA, Dozmorov M, Tang Y, Merrill JT, Wren JD, Sawalha AH. Genome-wide DNA methylation patterns in CD4+ T cells from patients with systemic lupus erythematosus. Epigenetics 2011; 6:593 - 601; http://dx.doi.org/10.4161/epi.6.5.15374; PMID: 21436623
- Etcheverry A, Aubry M, de Tayrac M, Vauleon E, Boniface R, Guenot F, Saikali S, Hamlat A, Riffaud L, Menei P, et al. DNA methylation in glioblastoma: impact on gene expression and clinical outcome. BMC Genomics 2010; 11:701; http://dx.doi.org/10.1186/1471-2164-11-701; PMID: 21156036
- Zawada AM, Rogacev KS, Hummel B, Grün OS, Friedrich A, Rotter B, Winter P, Geisel J, Fliser D, Heine GH. SuperTAG methylation-specific digital karyotyping reveals uremia-induced epigenetic dysregulation of atherosclerosis-related genes. Circ Cardiovasc Genet 2012; 5:611 - 20; http://dx.doi.org/10.1161/CIRCGENETICS.112.963207; PMID: 23074332
- Lübbert M, Tobler A, Daskalakis M. Cytosine demethylation of the proteinase-3/myeloblastin primary granule protease gene during phagocyte development. Leukemia 1999; 13:1420 - 7; http://dx.doi.org/10.1038/sj.leu.2401486; PMID: 10482994
- Zhang Q, Wang HY, Liu X, Wasik MA. STAT5A is epigenetically silenced by the tyrosine kinase NPM1-ALK and acts as a tumor suppressor by reciprocally inhibiting NPM1-ALK expression. Nat Med 2007; 13:1341 - 8; http://dx.doi.org/10.1038/nm1659; PMID: 17922009
- Ressler KJ, Mercer KB, Bradley B, Jovanovic T, Mahan A, Kerley K, et al. Post-traumatic stress disorder is associated with PACAP and the PAC1 receptor. Nature. England, 2011:492-7.
- Jung S, Yi L, Jeong D, Kim J, An S, Oh TJ, Kim CH, Kim CJ, Yang Y, Kim KI, et al. The role of ADCYAP1, adenylate cyclase activating polypeptide 1, as a methylation biomarker for the early detection of cervical cancer. Oncol Rep 2011; 25:245 - 52; PMID: 21109983
- Matsusaka K, Kaneda A, Nagae G, Ushiku T, Kikuchi Y, Hino R, et al. Classification of Epstein-Barr virus-positive gastric cancers by definition of DNA methylation epigenotypes. Cancer Res. United States, 2011:7187-97.
- Cogdell D, Chung W, Liu Y, McDonald JM, Aldape K, Issa JP, et al. Tumor-associated methylation of the putative tumor suppressor AJAP1 gene and association between decreased AJAP1 expression and shorter survival in patients with glioma. Chin J Cancer. China, 2011:247-53.
- Rauch TA, Wang Z, Wu X, Kernstine KH, Riggs AD, Pfeifer GP. DNA methylation biomarkers for lung cancer. Tumour Biol 2012; 33:287 - 96; http://dx.doi.org/10.1007/s13277-011-0282-2; PMID: 22143938
- Kim YH, Lee HC, Kim SY, Yeom YI, Ryu KJ, Min BH, Kim DH, Son HJ, Rhee PL, Kim JJ, et al. Epigenomic analysis of aberrantly methylated genes in colorectal cancer identifies genes commonly affected by epigenetic alterations. Ann Surg Oncol 2011; 18:2338 - 47; http://dx.doi.org/10.1245/s10434-011-1573-y; PMID: 21298349
- Tessema M, Yu YY, Stidley CA, Machida EO, Schuebel KE, Baylin SB, Belinsky SA. Concomitant promoter methylation of multiple genes in lung adenocarcinomas from current, former and never smokers. Carcinogenesis 2009; 30:1132 - 8; http://dx.doi.org/10.1093/carcin/bgp114; PMID: 19435948
- Ramos EA, Camargo AA, Braun K, Slowik R, Cavalli IJ, Ribeiro EM, Pedrosa FdeO, de Souza EM, Costa FF, Klassen G. Simultaneous CXCL12 and ESR1 CpG island hypermethylation correlates with poor prognosis in sporadic breast cancer. BMC Cancer 2010; 10:23; http://dx.doi.org/10.1186/1471-2407-10-23; PMID: 20109227
- Karouzakis E, Rengel Y, Jungel A, Kolling C, Gay RE, Michel BA, et al. DNA methylation regulates the expression of CXCL12 in rheumatoid arthritis synovial fibroblasts. Genes Immun. England, 2011:643-52.
- Yoshino M, Suzuki M, Tian L, Moriya Y, Hoshino H, Okamoto T, Yoshida S, Shibuya K, Yoshino I. Promoter hypermethylation of the p16 and Wif-1 genes as an independent prognostic marker in stage IA non-small cell lung cancers. Int J Oncol 2009; 35:1201 - 9; PMID: 19787276
- Zhou W, Jiang Z, Song X, Liu Y, Wen P, Guo Y, Xu F, Kong L, Zhang P, Han A, et al. Promoter hypermethylation-mediated down-regulation of CXCL12 in human astrocytoma. J Neurosci Res 2008; 86:3002 - 10; http://dx.doi.org/10.1002/jnr.21746; PMID: 18512766
- Wendt MK, Johanesen PA, Kang-Decker N, Binion DG, Shah V, Dwinell MB. Silencing of epithelial CXCL12 expression by DNA hypermethylation promotes colonic carcinoma metastasis. Oncogene 2006; 25:4986 - 97; http://dx.doi.org/10.1038/sj.onc.1209505; PMID: 16568088
- Sato N, Matsubayashi H, Fukushima N, Goggins M. The chemokine receptor CXCR4 is regulated by DNA methylation in pancreatic cancer. Cancer Biol Ther 2005; 4:70 - 6; http://dx.doi.org/10.4161/cbt.4.1.1378; PMID: 15662133
- Shen RZ, Pan S, Qi SJ, Lin XL, Cheng SD. Epigenetic repression of microRNA-129-2 leads to overexpression of SOX4 in gastric cancer. Biochem Biophys Res Commun 2010; 394:1047 - 52; http://dx.doi.org/10.1016/j.bbrc.2010.03.121; PMID: 20331975
- Reinert T, Modin C, Castano FM, Lamy P, Wojdacz TK, Hansen LL, Wiuf C, Borre M, Dyrskjøt L, Orntoft TF. Comprehensive genome methylation analysis in bladder cancer: identification and validation of novel methylated genes and application of these as urinary tumor markers. Clin Cancer Res 2011; 17:5582 - 92; http://dx.doi.org/10.1158/1078-0432.CCR-10-2659; PMID: 21788354
- Sugino Y, Misawa A, Inoue J, Kitagawa M, Hosoi H, Sugimoto T, Imoto I, Inazawa J. Epigenetic silencing of prostaglandin E receptor 2 (PTGER2) is associated with progression of neuroblastomas. Oncogene 2007; 26:7401 - 13; http://dx.doi.org/10.1038/sj.onc.1210550; PMID: 17533365
- Isidoro-García M, Sanz C, García-Solaesa V, Pascual M, Pescador DB, Lorente F, Dávila I. PTGDR gene in asthma: a functional, genetic, and epigenetic study. Allergy 2011; 66:1553 - 62; http://dx.doi.org/10.1111/j.1398-9995.2011.02685.x; PMID: 21883277
- Yi JM, Dhir M, Guzzetta AA, Iacobuzio-Donahue CA, Heo K, Yang KM, Suzuki H, Toyota M, Kim HM, Ahuja N. DNA methylation biomarker candidates for early detection of colon cancer. Tumour Biol 2012; 33:363 - 72; http://dx.doi.org/10.1007/s13277-011-0302-2; PMID: 22238052
- Koch CM, Wagner W. Epigenetic-aging-signature to determine age in different tissues. Aging (Albany NY) 2011; 3:1018 - 27; PMID: 22067257
- Tao R, Li J, Xin J, Wu J, Guo J, Zhang L, Jiang L, Zhang W, Yang Z, Li L. Methylation profile of single hepatocytes derived from hepatitis B virus-related hepatocellular carcinoma. PLoS One 2011; 6:e19862; http://dx.doi.org/10.1371/journal.pone.0019862; PMID: 21625442
- Arai E, Chiku S, Mori T, Gotoh M, Nakagawa T, Fujimoto H, Kanai Y. Single-CpG-resolution methylome analysis identifies clinicopathologically aggressive CpG island methylator phenotype clear cell renal cell carcinomas. Carcinogenesis 2012; 33:1487 - 93; http://dx.doi.org/10.1093/carcin/bgs177; PMID: 22610075
- Oka D, Yamashita S, Tomioka T, Nakanishi Y, Kato H, Kaminishi M, Ushijima T. The presence of aberrant DNA methylation in noncancerous esophageal mucosae in association with smoking history: a target for risk diagnosis and prevention of esophageal cancers. Cancer 2009; 115:3412 - 26; http://dx.doi.org/10.1002/cncr.24394; PMID: 19472401
- Oster B, Thorsen K, Lamy P, Wojdacz TK, Hansen LL, Birkenkamp-Demtröder K, Sørensen KD, Laurberg S, Orntoft TF, Andersen CL. Identification and validation of highly frequent CpG island hypermethylation in colorectal adenomas and carcinomas. Int J Cancer 2011; 129:2855 - 66; http://dx.doi.org/10.1002/ijc.25951; PMID: 21400501
- Morris MR, Ricketts CJ, Gentle D, McRonald F, Carli N, Khalili H, Brown M, Kishida T, Yao M, Banks RE, et al. Genome-wide methylation analysis identifies epigenetically inactivated candidate tumour suppressor genes in renal cell carcinoma. Oncogene 2011; 30:1390 - 401; http://dx.doi.org/10.1038/onc.2010.525; PMID: 21132003