Abstract
Enhancers are cis-acting elements capable of regulating transcription in a distance and orientation-independent manner. A subset of enhancers are occupied by RNA polymerase II (RNAP II) and transcribed to produce long non-coding RNAs termed eRNAs. We thoroughly investigated the association between eRNA productivity and various chromatin marks and transcriptional regulators in mouse embryonic stem cells (ESCs) through an integrative approach. We found that eRNA-producing enhancers exhibited elevated levels of the active mark H3K27Ac, decreased DNA methylation, and enrichment for the DNA hydroxylase Tet1. Many eRNA-producing enhancers have recently been characterized as “super-enhancers,” suggesting an important role in the maintenance of pluripotency. Using experimental methods, we focally investigated a well-characterized enhancer linked to the Nanog locus and confirmed its exclusive eRNA productivity in ESCs. We further demonstrate that the binding of Sall4 and Tet family proteins were required for eRNA productivity at this locus. Collectively, we demonstrate that Tet1 binding and DNA hypomethylation are hallmarks of eRNA production.
Introduction
Two types of genetic elements mediate transcriptional regulation: trans-acting factors, typically proteins that regulate transcription by binding to cis-acting elements, which are DNA regulatory elements on the same contiguous chromatin segment as the regulated locus. The study of cis-regulatory elements has previously focused on promoters, which regulate transcription in a distance and orientation-dependent manner. In contrast, enhancers are cis-regulatory elements that act essentially independently of distance and orientation, and are largely responsible for lineage restricted gene expression.Citation1 Recent work from the ENCODE project indicates that a large fraction of the genome that was previously considered “junk” DNA in fact contains a variety of cis-regulatory elements, including enhancers.Citation2,Citation3
Seminal work from Bing Ren’s group initially described a series of epigenetic marks that could be used to identify enhancer elements.Citation4-Citation6 Since then, a variety of marks and/or combinations have been described. Monomethylation at histone 3 Lysine 4 (H3K4me1) and binding by the histone acetyltransferase p300 /CBP were the first described marks, with more recent work illustrating that acetylation of H3K27 is a reliable mark of “active” vs. “poised” enhancers.Citation4,Citation7-Citation9 In addition, the binding of core groups of lineage specific transcription factors (TFs) has been used to define enhancers.Citation10 However, the biological differences between these alternative classification strategies remains unclear.Citation2,Citation11,Citation12
A subset of enhancers, defined by H3K4me1 enrichment and p300 occupancy, are bound by RNA polymerase II (RNAP II) and transcribed to produce a novel class of non-coding RNAs (ncRNAs), termed eRNAs (enhancer transcribed RNAsCitation1,Citation13-Citation16). These RNAs are typically bidirectionally transcribed (i.e., both strands of the enhancer), unspliced, and considered “long” ncRNAs (as they are greater than 1kb in size).Citation17 eRNAs seem to assist in enhancer function, perhaps by tethering enhancer-critical proteins to chromatin, but a number of fundamental questions remain.Citation18-Citation20 For example, what distinguishes transcribed vs. non-transcribed enhancers beyond RNAP II occupancy? In addition, given the different epigenetic “definitions” of enhancers, is eRNA production ubiquitous among all types of enhancers, or specific to chromatin regions defined by certain epigenetic marks?
It is technically challenging to fully address these questions, since this requires the identification of rare transcripts (eRNAs). Since these RNAs must be identified based upon their production from enhancers, it also requires the ability to systematically identify different classes of distal cis-regulatory elements while simultaneously distinguishing them from other ncRNAs as well as unannotated genes. For example, long-intergenic non-coding RNAs (lincRNAs) may play a role as scaffolding molecules to allow epigenetic regulators to interact with chromatin.Citation21 Unlike eRNAs however, they are typically spliced and transcribed from loci with similar epigenetic marks as protein-coding loci, namely H3K4me3-rich at their promoters and H3K36me3-rich in the transcribed segment(s).Citation22 Thus, a comprehensive “atlas” of the epigenetic landscape of a single lineage is required for the proper identification of eRNAs. In a mammalian system, many cell types have been investigated to answer these types of questions, but mouse embryonic stem cells (ESCs) have been studied in sufficient details so that a comprehensive map of their transcription factor (TF) binding sites and epigenetic marks has been developed.Citation23 ESCs are derived from the inner cell mass of embryos and possess two canonical properties: self-renewal, the ability to propagate indefinitely in an undifferentiated state, and pluripotency, the ability to differentiate into all three primitive germ layers (mesoderm, endoderm, and ectoderm; reviewed by YoungCitation24). Both properties are controlled at the level of transcription by a group of lineage-specific TFs, including Nanog, Oct4, Sox2, and Sall4, among others.Citation25-Citation27
Our goal was to harness existing ChIP-seq and transcriptome data sets to comprehensively identify any distinguishing epigenetic marks and/or protein binding events associated with eRNA producing enhancers, which consist of only a small subset of all enhancers. Surprisingly, we also found that the enhancers transcribed in ESCs alone exhibited lower overall DNA methylation and were occupied by the Tet family of DNA hydroxylases.
Results
Comprehensive identification and comparison of enhancers within ESCs
The initial studies on eRNA production focused on a specific type of enhancer-regions (at least 1 kb in size) enriched on H3K4me1 and co-occupied by p300/CBP.Citation13-Citation15 Since these initial studies, additional enhancer definitions have been described, such as H3K27Ac being a useful mark to identify “active” enhancers. An additional definition for tissue-specific enhancers is extragenic sites co-occupied by multiple lineage specific transcription factors. Excellent work has shown in ESCs that these sites, which are co-occupied by the master transcription factors Nanog, Oct4, Sox2, and components of the mediator complex (abbreviated NOSM), collectively form “super-enhancers” that regulate pluripotency-specific genes.Citation10,Citation28 Collectively, these results highlight the evolving understanding of enhancers. To date, there has yet to be a consensus set of markers for enhancer activity.
As a first step, we comprehensively analyzed and compared eRNA producing enhancer signatures based upon four different, commonly used criteria (H3K4me1 positive, H3K27Ac positive, NOSM, and p300-occupied). Published ChIP-seq data sets were obtained for a variety of transcription factors (TFs), histone modifications, RNA polymerases, and other regulatory proteins (Table S1). To identify regions bound by these factors, all data sets were re-analyzed using a uniform bioinformatics pipeline. Peak-calling was determined by using a highly stringent P value (see Methods section for details) to reduce false-positives. To eliminate incorrect enhancer annotations due to similarity with alternate promoters, or the promoters of un-annotated genes, pseudogenes, and/or lincRNAs, any peaks that overlapped with genomic regions rich in H3K4me3 were removed. While H3K4me3 is classically thought of as a promoter mark, at least one group has reported it also marks active enhancers in T cells.Citation29 Removal of H3K4me3 enriched extragenic regions may therefore remove a group of potent enhancers. Any sites that overlapped with extragenic regions enriched with H3K36me3 were also removed; this step was not performed on intragenic sites due to possible overlap between exonic enhancers and H3K36me3. This gave us four “groups” of enhancers based upon different definitions (). In general, the definitions were not mutually exclusive, but a large fraction was unique (). RNAP II occupied a minority of enhancers (<25%, ). Overall, the elongating form of RNAP II (Serine 2 phosphorylated, Ser2p) was infrequently observed at enhancers (data not shown). In contrast, the initiating form of RNAP II (Serine 5 phosphorylated, Ser5p), or an antibody against all RNAP II species, frequently detected RNAP II occupancy at enhancers. RNAP II Ser5p is typically found at promoters and is often associated with bidirectional transcription,Citation30-Citation32 consistent with the pattern of transcription identified at enhancers.Citation15,Citation16,Citation33,Citation34 To identify eRNA-producing enhancers, a published data set of genome-wide nuclear run-on followed by next-generation sequencing (GRO-seq) was obtained.Citation31 This approach maps the production of nascent RNA molecules from transcriptionally engaged RNAP II, and has been used previously to identify eRNAs.Citation13 Using this data set, a larger percentage of enhancers were transcribed (approximately 50%) than would be expected based upon our RNAP II data. This may be related to the stringent P-value cutoff (10−6) used to identify RNAP II binding sites, thereby increasing the number of false-negative genomic regions within our data set. However, we cannot exclude that some of these regions may be transcribed by non-RNAP II RNA polymerases. Nonetheless, of the regions bound by RNAP II, the majority (>75%) exhibited some degree of transcription. In addition, GRO-seq was also performed on mouse embryonic fibroblasts (MEFs), which allowed the identification of enhancers that were transcribed in a cell-type specific manner. A small group of enhancers (6–10%) within each class were bound by RNAP II and produced eRNAs in ESCs alone. In contrast, a much larger group of enhancers were transcriptionally silent in both ESCs and MEFs. Given the strong differences between the enhancers transcribed specifically in ESCs (hereafter referred to as ESC-specific enhancers) and those that produced no eRNA in either ESCs or MEFs (hereafter referred to as eRNA-negative enhancers), we utilized this distinction to further explore differences in eRNA production. As a first step, we used our overlap procedure to determine whether eRNA negative or ESC-specific enhancers showed significant overlap among the different definitions of enhancers. As can be seen in , the ESC-specific enhancers showed higher overlap among the enhancer definitions than eRNA negative enhancers. This implies that ESC-specific enhancers share multiple epigenetic characteristics, indicating they are likely highly active.
Table 1. A listing of the numbers and percentage for each class of enhancers in different classes
Figure 1. (A) Four-way Venn diagram to indicate the overlap between the four different classes of enhancers used in this study. Red numbers indicate the percent of enhancers that were unique to a given definition. (B) The different enhancer classes were linked to their nearest neighbor gene, and expression of the different gene sets was assessed by GSEA, comparing ESCs at day 0 and day 6-post retinoic acid differentiation. ESC-specific and eRNA-negative enhancers are defined in the text. Numbers within each box indicate the normalized enrichment score, defined by GSEA. Details on how this score is calculated are provided in the original manuscript describing GSEA.Citation38 Plus scores indicate a correlation of the gene-set with the Day 0 time point, Minus signs would indicate a correlation with the Day 6 time point. Red indicates a statistically significant (P < 0.05 and FDR < 10%) correlation with the Day 0 time point, black indicates the NES score was not statistically significant.
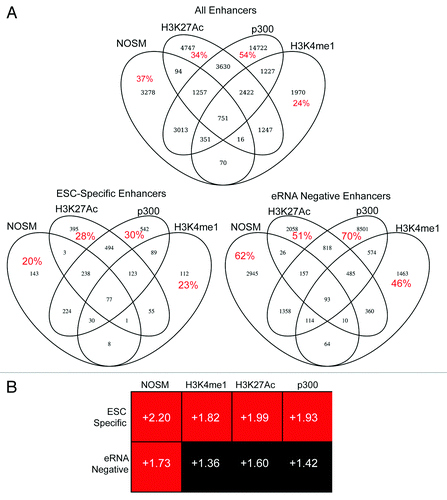
Next, each enhancer was mapped to the nearest gene as a putative target. The nearest neighbor analysis has been used previously to identify enhancer:gene pairs and tends to show high correlation with other methods,Citation28 although it is not nearly as comprehensive as utilizing chromosomal conformational capture based approaches.Citation2,Citation11,Citation12,Citation35-Citation37 We then assessed whether genes linked to either ESC-specific or eRNA-negative enhancers were enriched in pluripotent cells. We used Gene Set Enrichment Analysis (GSEACitation38) on a published microarray data set of ESCs differentiated by exposure to retinoic acid (RA), which causes a rapid, uniform loss of pluripotency.Citation25,Citation27 GSEA determines whether a list of genes (termed “gene set”) are enriched for expression level changes between two conditions: in our case, pluripotent ESCs vs. differentiated (RA-exposed) cells. The primary output is a normalized enrichment score, in which the relative enrichment of the gene set in either of the two microarray data sets is calculated and then normalized for differences in gene set size.Citation38 In general, the ESC-specific enhancer-linked genes showed higher normalized enrichment scores in ESCs than in RA-differentiated cells (). This difference was statistically significant (P < 0.01 and FDR < 10%) for some of the enhancer classes (H3K4me1, H3K27Ac, p300), although for NOSM enhancers both the ESC-specific and eRNA-negative enhancer-linked genes showed a statistically significant enrichment in ESCs as opposed to RA-differentiated cells. This is not surprising, given that RA-differentiated cells do not express Nanog, Oct4, or Sox2.Citation25,Citation27 Collectively, these results indicate that the target genes are enriched in ESCs compared with differentiated cells.
eRNA production rates are RNAP II-driven and independent of promoter transcription
Next, we wanted to determine if there were differences in eRNA production between the different enhancers. To assess eRNA production at each of our different classes, genomic regions were split into extragenic and intragenic enhancers, and for intragenic enhancers only antisense strand transcription was analyzed. As can be seen in Figure S1A , for each class of enhancers, RNAP II-bound enhancers showed higher eRNA production rates (Mann-Whitney test, P < 10−16) than unbound regions. ESC-specific enhancers for each class showed the same degree of transcription as other RNAP II-bound enhancers, implying that cell-type specificity in eRNA production rates did not correlate with higher enhancer transcriptional activity. We next directly compared eRNA production from the different classes of enhancers (). Overall, the median eRNA production rate from each group of enhancers occupied by RNAP II and ESC-specific enhancers is highly similar. Collectively, this implies that eRNA production occurs at enhancers defined by different criteria at similar rates.
Figure 2. Box Plots to show relative transcription rates (RPKM) for RNAP II Bound and enhancers that produce an eRNA in an ESC specific fashion. Outliers are omitted from the graph.
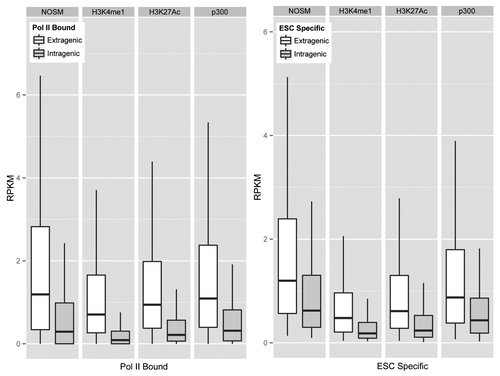
To determine if enhancer and promoter transcription rates are correlated, the GRO-seq data set was re-analyzed at well-annotated promoters. GRO-seq, by design, measures transcriptional rates, rather than transcript levels, similar to nuclear run-off assays. For each of the different classes of enhancers, we found no correlation between the enhancer transcriptional rates (RPKM) and those of its nearest promoter, regardless of class (Fig. S1B). Thus, enhancer transcription is likely dependent on RNAP II, but enhancers of different definitions show overall equivalent levels of transcription, and promoter transcription rates are not predictive of enhancer transcription rates. Given that our enhancers were within relatively close proximity to their assigned genes (no more than 50 kb), it is likely that enhancer transcription is not simply due to these chromatin segments being transcribed at a higher rate, or eRNA production being a byproduct of higher promoter transcriptional rates. Instead, it appears that promoter- and enhancer-transcription are regulated independently.
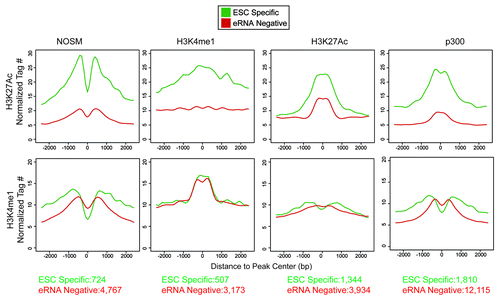
Recent work has shown that H3K27Ac and H3K4me1 can be used to mark active (H3K4me1+/H3K27Ac+) vs. poised (H3K4me1+/H3K27Ac−) enhancers, implying that H3K27Ac correlates with enhancer activity.Citation7,Citation9 We assessed levels of both histone marks at ESC-specific and eRNA-negative enhancers (). We found that H3K4me1 levels were similar between ESC-specific and eRNA-negative enhancers. In contrast, H3K27Ac was substantially higher at ESC-specific enhancers, indicating that eRNA production represents a more active subset of enhancers.
Figure 3. Profiles of the histone methylation status (H3K4me1) or acetylation (H3K27Ac) for enhancers of different definitions. X-axis is the distance away from center of the enhancers in a 5 kb window. Y-axis is the normalized tag number for either H3K27Ac or H3K4me1 ChIP. The number of enhancers present in each category is presented at the bottom of the figure.
eRNA-producing enhancers are hypomethylated and occupied by Tet1
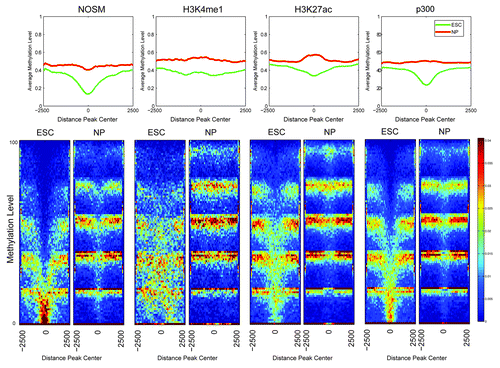
Seminal work in the rapidly evolving field of DNA methylation has revealed that distal cis-regulatory elements, including enhancers, showed lower overall levels of DNA methylation.Citation39 In this publication, a Hidden Markov Model type approach determined that enhancers exhibit low levels of methylation (LMR), which is an intermediate between unmethylated (unmethylated region, UMR) and fully methylated (fully methylated region, FMR). Of note, the method used (BiSeq) for genome-wide methylation analysis cannot distinguish between methylcytosine and hydroxymethylcytosine. Lack of methylation at CpGs within promoter elements (UMRs) correlates with mRNA transcription, and we hypothesized that a similar DNA methylation pattern would be present at enhancers. As a first step, the degree of DNA methylation for the different classes of enhancers was determined, separated by whether they were eRNA-negative or ESC-specific manner () in a 5kb window around the center of each enhancer. As can be seen, overall there was a “shift” in that ESC-specific eRNA producing enhancers tended to show a lower level of methylation than eRNA-negative enhancers in the same class. Most surprising was that, while all enhancers exhibit low DNA methylation, many of the ESC-specific eRNA producing enhancers were substantially hypomethylated (peak center methylation < 20%). H3K4me1 marked enhancers did not exhibit as strong a degree of hypomethylation as the other classes. In general, ESC-specific enhancers showed more overlap with UMRs and LMRs (UMRs > LMRs) than either eRNA negative or all enhancers of a given definition (Fig. S2A), consistent with our earlier analysis (). One possibility is that the degree of methylation may simply relate to differences within the local genomic region, i.e., that eRNA-producing enhancers are located within chromatin regions with lower levels of DNA methylation regardless of cell-type. To address this, a similar analysis on DNA methylation status in ESC-derived neural progenitors (NPs) was performed.Citation39 Similar to the original paper, regions of DNA hypomethylation in the two cell types were lineage-specific, with the ESC-specific enhancers showing lower overall levels of DNA methylation in ESCs, but converting to more fully methylated regions after differentiation into a different cell type, indicating that eRNA-producing enhancers are capable of becoming FMRs (). Collectively, DNA hypomethylation provides an independent discrimination between transcriptionally active vs. inactive enhancers.
Figure 4. Heatmaps and profiles showing the average methylation levels for the different classes of enhancers, comparing ESC-Specific and eRNA-negative. The x-axis in both profiles and heatmaps corresponds to a 5 kb window centered at each enhancer. The y-axis is the average methylation level, in particular each row in the heatmap correspond to a different methylation level. The colors used in the heatmaps (from blue to red) represents the number of regions with a particular methylation level, with a color scheme legend on the far right of the lower panel.
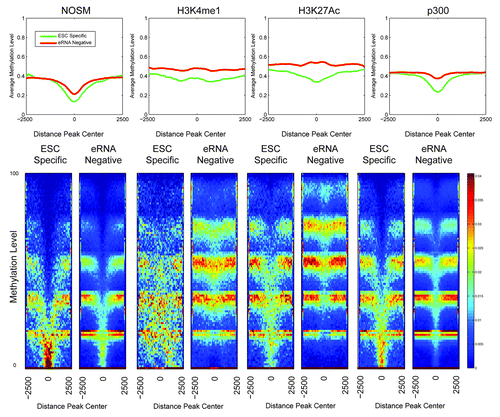
Figure 5. Heatmaps and profiles showing the average methylation levels comparing the enhancer regions in ESC and NP cells. The x-axis in both, the profiles and heatmaps, corresponds to a 5kb window centered at each enhancer. The y-axis is the average methylation level, in particular each row in the heatmap correspond to a different methylaton level. The colors used in the heatmaps (from blue to red) represents the number of regions with a particular methylation level, with a color scheme legend on the far right of the lower panel.
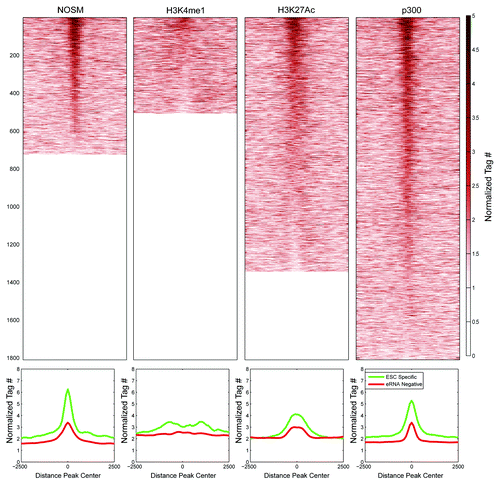
Given the correlation of reduced DNA methylation at enhancers and eRNA production, we hypothesized that Tet proteins would selectively bind ESC-specific enhancers over eRNA-negative ones.Citation39,Citation40 Tet1 occupancyCitation40 was determined at ESC-specific eRNA-producing and eRNA-negative enhancers. Tet1 was clearly enriched at NOSM, H3K27Ac, and p300 ESC-specific enhancers over eRNA-negative ones (; Fig. S2B). Surprisingly, H3K4me1 positive ESC-specific enhancers showed a smaller enrichment for Tet1 binding, which is consistent with their overall higher DNA methylation. This correlation between eRNA production, hypomethylation, and Tet1 occupancy is highly similar to what is seen at promoters, perhaps indicating a shared biological mechanism for transcription of promoters and enhancers, at least in ESCs.
Figure 6. Profiles and heatmaps showing the normalized Tet1 signal for different classes of enhancers: ESC-Specific and eRNA-negative. The x-axis in both profiles and heatmaps corresponds to a 5kb window centered at each enhancer. The y-axis in the profiles reports the average normalized tag counts for all regions considered. Each row in the heatmap corresponds to one ESC-specific enhancer region.
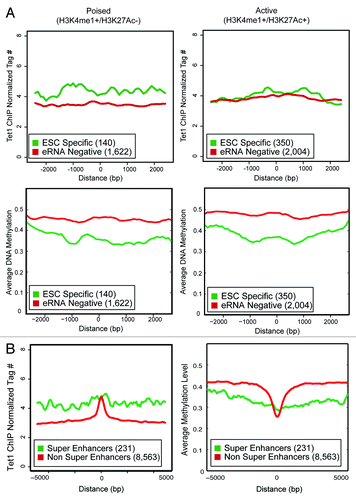
Given recent interest in identifying active or lineage-critical enhancers,Citation7,Citation9,Citation28 we queried whether further subdividing enhancers based upon histone modifications into active (H3K4me1+/H3K27Ac+) or poised (H3K4me1+/H3K27Ac−) would reveal differences in DNA methylation and Tet1 occupancy. We observed that ESC-specific enhancers, whether active or poised, showed lower DNA methylation than their eRNA-negative counterparts (). Similar to recently published work,Citation41 Tet1 occupancy was higher at poised, ESC-specific enhancers than others. Collectively, this indicates that Tet1 occupancy is not simply a result of active enhancers displaying higher levels of H3K27Ac. Recent work utilizing higher coverage next-generation sequencing also reveals that among NOSM enhancers, a small number (231 out of 8794) were considered “super-enhancers” because they exhibited higher levels of Mediator and were typically larger (≈10 kb) than classical transcriptional enhancers.Citation28 These super-enhancers were both highly active and extremely lineage restricted. We found that super enhancers were predominantly RNAP II-occupied (92%), and 56% were ESC specific. In contrast, among the 8563 non-super enhancers, a minority (19%) were RNAP II-occupied, and 11% were ESC specific, illustrating that the super enhancers and ESC-specific enhancers are functionally similar. Super enhancers exhibited a higher level of genomic-occupancy by Tet1 and lower DNA methylation over a large region of chromatin (>5 kb), although the peak of binding by Tet1 was typically higher for non-super enhancers in a small region (≈1 kb) around the center (). A similar observation was found in the original publication,Citation28 with transcription factors (Nanog, Oct4, Sox2) and Mediator components (Med1/12) present on a large (approximately 5–10 kb) region of chromatin, rather than having a single, distinct peak over a smaller region. Thus, ESC-specific enhancers are enriched for lineage-restricted, highly active enhancers.
Figure 7. (A) The Tet1 occupancy and average DNA methylation for active and poised enhancers is shown over a 5 kb window. Y-axis is either normalized tag count (Tet1) or average DNA methylation. X-Axis is distance from the center of the enhancer. The number of enhancers present in each category is indicated in parentheses. (B) The Tet1 occupancy and average DNA methylation for super and non-super enhancersCitation28 is shown over a 10 kb window. A larger window was used because super-enhancers tend to be larger than typical enhancers. Y-axis is either normalized tag count (Tet1) or average DNA methylation. X-Axis is distance from the center of the enhancer. The number of enhancers present in each category is indicated in parentheses.
A Nanog-linked enhancer produces an eRNA in a Tet-dependent manner
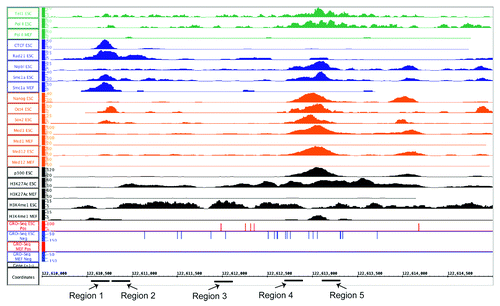
To better address the mechanism behind eRNA production, we chose to identify a known enhancer linked to a pluripotency-critical locus. Immediately 45 kb upstream of the Nanog locus is a previously described enhancer elementCitation42 originally identified by differential DNase I hypersensitivity (DHS). This same region overlapped, at least partially, with all four classes of enhancers (). Nanog, Oct4, Sox2, and both Med1 and Med12 occupy this enhancer in ESCs, and Med1 and Med12 exhibit no binding in MEFs. The region shows a strong signal for both H3K4me1 and H3K27Ac, and has a p300-binding site as well, which is not observed in MEFs. In terms of RNAP II binding, there is robust binding that overlaps with the NOS site, which was also determined in the original publication and is not observed in MEFs. To verify our genomic data sets, we queried ENCODE-derived data sets available through the UCSC genome browser and found that this region exhibits ESC specific H3K4me1, and in ESCs are occupied by RNAP II and p300 (data not shown). In addition, this region has two distinct LMRs in ESCs that become fully methylated in NPs (Fig. S3A).
Figure 8. An IGB Screen Capture of the enhancer region approximately 45kb upstream of the Nanog locus which produces an eRNA. Normalized wiggle files are shown for all the factors analyzed, as well as the GRO-seq data utilized for identifying eRNA production in ESCs and MEFs. For ChIP-seq data sets, y-axis shows normalized tag counts. For GRO-seq data sets, it is RPKMs for the plus (red) or minus (blue) strands. For all, the genomic position (mm9) is indicated on the x-axis. The PCR primer regions amplified for various qPCR assays are indicated below the x-axis. For signals where both ESC and MEF are displayed, the Y-axis for signals in the two cells lines are the same scale.
To confirm this region contains enhancer potential, a 2.8 kb fragment of this region was cloned downstream of a firefly-derived luciferase reporter. To drive luciferase expression either a minimal SV40 derived promoter, or a 1.5 kb fragment of the endogenous Nanog promoter was utilized. Clear enhancement of luciferase in the plasmids with the enhancer in either orientation as compared with the same reporter without any enhancer in ESCs was observed (Fig. S3B). Transfecting the same reporters into a mouse fibroblast cell line (3T3) revealed no change from baseline in luciferase activity, confirming this enhancer is ESC-specific. In addition to the cell-type specificity, there was a consistently lower level of luciferase enhancement when the enhancer was cloned in the opposite orientation (Minus) regardless of the promoter used. While typically enhancers are orientation independent, given the relatively large size of this enhancer (≈3 kb) as opposed to most studies, this may simply represent difficulties with DNA bending when the backbone (5.5 kb) is less than twice the size of the enhancer element (5.6 kb). Alternatively, there may be a cryptic insulator within this region, which was not detected by ChIP-seq. Nonetheless, this DNA element is an ESC-specific transcriptional enhancer.
Next, we wanted to confirm that eRNAs are produced from this locus in an ESC-specific manner. Given the debate in the literature as to whether eRNAs are polyadenylated, two different priming methods for cDNA generation were used. RNA was harvested from ESC exposed to RA at different time points, and reverse transcription reactions (RT) were primed with either Oligo dT or random hexamers. Quantitative real-time PCR was performed using two independent primer sets to the eRNA produced by this enhancer and, as a control, primers to Nanog mRNA were used as well (). Both sets of primers to the eRNA detected transcript around the RNAP II binding site. As the ESCs were differentiated over a period of 6 d, there was a rapid decline in Nanog mRNA and the eRNA. An almost identical pattern was observed from two distinct primer sets approximately 1 kb 5′ of the RNAP II site, implying the enhancer is transcribed over a relatively long distance (data not shown), as has been observed by others.Citation13,Citation14
Figure 9. (A) Nanog mRNA and eRNA levels using different priming methods after exposing ESCs to retinoic acid (RA). Two primers for the eRNA are shown to reduce the likelihood that a spurious species was being detected. Error bars represent SEM of three experiments. (B) Metabolically labeled versions of Nanog, Sall4a, and Sall4b,which have been described previously, had bioChIP performed, followed by quantitative PCR using primers specific to different regions (x-axis). Fold-enrichment relative to sheared genomic DNA (Input) is indicated on the y-axis. A region that exhibits minimal occupancy by pluripotency factors in ESCs was included (negative control). Error bars represent SEM of three experiments. (C) Antibody-mediated ChIP followed by qPCR was performed over the same region using the same primers, with antibodies to Tet1, Tet2, or RNA Polymerase II. As a control, a nonspecific IgG and an antibody to a core subunit of RNA Polymerase III (RPC32) were included. Error bars represent SEM of three experiments.
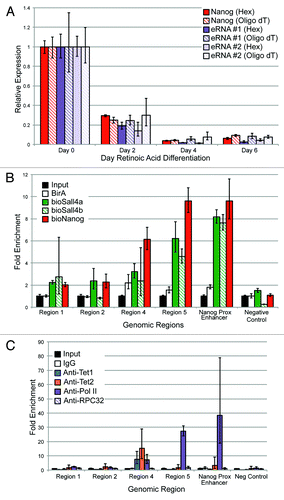
To confirm that known pluripotency factors bind to this enhancer, we used our previously published metabolic labeling approachCitation25,Citation43 with cells expressing biotinylatable versions of Nanog, and two isoforms of the known pluripotency TF Sall4 (Sall4a and Sall4b, ). As predicted by ChIP-seq, Nanog binds to this enhancer at the expected location, and also bound both isoforms of Sall4. As a positive control, binding by Nanog and both Sall4 isoforms to a well-described enhancer approximately 5 kb upstream of the Nanog TSS was verified; this region has been proposed by others to also represent an alternative promoter/TSS for Nanog.Citation44,Citation45 The Nanog-proximal enhancer was removed from our original analysis because it displays high levels of H3K4me3, consistent with the fact that it may have promoter activity. Other regions within the enhancer did not appear to bind either Sall4 isoform or Nanog (Regions 1 and 2), whereas the region immediately adjacent (Region 4) displayed a lower level of occupancy. Antibody-based ChIP-qPCR was performed to verify Tet1 and RNAP II binding, and also to determine if Tet2 could occupy this region. Both Tet1 and Tet2 bound to a single region (, Region 4) within the enhancer, consistent with the ChIP-seq data for Tet1. RNAP II exhibited binding to both Region 4 and Region 5, consistent with the ChIP-seq data seen in this region. No binding was detected using a non-specific antibody (IgG) or an antibody to RPC32 (a core subunit of RNAP III) to any of the elements within the enhancer, the Nanog proximal enhancer, or a genomic region within a gene desert (negative control).
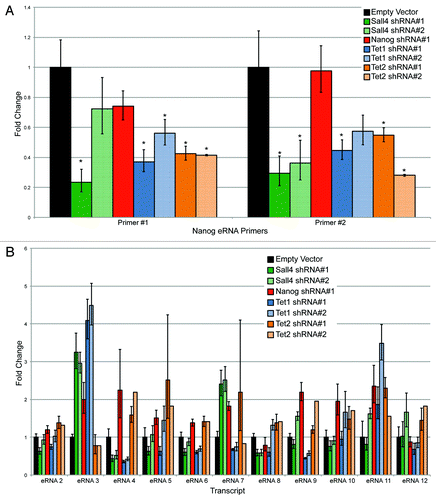
Given that Tet1, Tet2, Nanog, and Sall4 bound to this region immediately adjacent to the RNAP II binding site, we wanted to determine the effect of depleting these factors on eRNA production. ESCs were infected with lentiviruses containing two different shRNAs to Sall4, Nanog, Tet1, and Tet2. Cells were selected after two rounds of lentiviral infection with puromycin to enrich for a high multiplicity of infection. After 48 h, puromycin-treated cells were collected. A relatively early time point was used to ensure that the ESCs have not yet begun to differentiate based upon morphology (ref. Citation25 and data not shown). As seen in Figure S4A, the shRNAs induced a substantial reduction in mRNA levels, with an almost complete depletion of the protein as well (Fig. S4B). The second shRNA to Nanog (shRNA #2) yielded suboptimal knockdown (<50%), and is not shown for subsequent analyses, although it gave similar results to shRNA #1 (data not shown). The pluripotency marker Oct4, showed a modest reduction at the mRNA and protein levels with Nanog or Sall4 depletion, and to a lesser extent Tet1. Tet1 depletion caused a modest reduction in Tet2 (Fig. S4). The regulation of Oct4 and Nanog by Sall4 is well known,Citation25,Citation46 and our results indicate that Sall4 likely regulates the expression of both Tet1 and Tet2. There was a consistent reduction in the levels of Nanog transcript after Tet1 depletion, but not Tet2, as seen previously by others.Citation47 Two different primer sets to detect the eRNA were used to ensure that spurious PCR products were not being detected. Depletion of Sall4 or either shRNA to Tet1 or Tet2 reduced the eRNA to 30–60% of endogenous levels (). Surprisingly, Nanog depletion caused minimal reductions in eRNA level. The reduction in eRNA levels was not due to a global lowering of RNAP II levels (Fig. S4B).
Figure 10. (A) Assessing eRNA production using two independent primer sets after shRNA mediated depletion by various factors. An asterisk indicates P < 0.05 difference from the empty vector control using a Student’s T test. (B) Assessment of eRNA levels after shRNA mediated depletion of Sall4, Nanog, Tet1, or Tet2 from an additional 11 (12-total) enhancers.
Given the clear correlation between eRNA production and DNA hypomethylation on a genome-wide basis (), we wanted to investigate whether Sall4, Nanog, and Tet1 were directly responsible for maintaining the Nanog-linked enhancer in a hypomethylated state. ESCs were infected with a single shRNA for each protein and the empty lentiviral vector, and bisulfite sequencing performed (Fig. S5). All CpGs (for a total of 41) were sequenced in an approximately 1.8 kb region of the Nanog-linked enhancer that overlapped the binding sites of Sall4, Nanog, RNAP II, and Tet1, and contains a single CpG island (shown in red). As expected, in ESCs infected with the empty vector very little CpG methylation was observed (<5%). Surprisingly, there was essentially no change in CpG methylation with depletion of Sall4, Nanog, or Tet1. These results indicate that enhancer DNA hypomethylation is not dependent upon binding of either pluripotency-associated TFs (Nanog, Sall4) or Tet1 alone.
Collectively, these data demonstrate that the Nanog-linked enhancer produces an eRNA in an ESC specific fashion, is occupied by known pluripotency-associated TFs such as Nanog and Sall4, but also occupied by both Tet1 and Tet2. Depletion of either Tet protein caused a statistically significant change in eRNA levels, showing that enhancer transcription at this locus is partially dependent on Tet proteins.
Additional eRNA producing enhancers show a variable dependence on TFs and Tet proteins
Depletion of TFs and especially Tet proteins has variable effects on promoter transcription, with many genes being activated or repressed, indicating that these factors have pleiotropic effects on RNA production.Citation25,Citation40,Citation48,Citation49 To determine if the effects on eRNA production observed at the Nanog −45 kb enhancer were a more global phenomenon, we identified 11 additional ESC-specific (eRNAs 2–12), Tet1 occupied enhancers, and measured eRNA levels after shRNA depletion of Sall4, Nanog, Tet1, and Tet2. We found a highly variable response (; genomic regions specified in Table S4). For example, eRNA #2 showed minimal changes in its levels after depletion of Sall4, Nanog, Tet1, or Tet2. In contrast, eRNA #4 was reduced after depletion for Sall4 or Tet1, but elevated after depletion of Nanog or Tet2. While some of these effects may be indirect after depletion of each factor, they point to the similarity between enhancers and promoters in their variable response after loss of either transcription factors or Tet1/2.
Discussion
Our work sheds light on the transcription of enhancers to produce eRNAs. Our first step was to identify and characterize enhancers in ESCs using a variety of different criteria to not only delineate which genomic regions possess enhancer potential, but also to systematically exclude other types of genomic features. Excellent work from other labsCitation22 clearly demonstrates multiple differences between eRNAs and lincRNAs. Specifically, the exclusion of genomic regions with H3K4me3 or H3K36me3 eliminates the possibility of lincRNAs falsely confounding our characterization of eRNAs. Like others, we found that eRNAs were produced typically in a bidirectional fashion (data not shown), consistent with occupancy by RNAP II Ser5p and not RNAP II Ser2p.Citation30 Collectively, this points to another classification difference that can be used to identify eRNA producing enhancers, i.e., regions bound by the initiating- but not elongating-form of RNAP II.
The fact that ESC-specific enhancers are more tightly linked to pluripotency-associated genes () is not surprising, given that our linking strategies used a distance-based method, similar to others, and that roughly half of the enhancers were intragenic. Thus, the ESC specific transcription of the gene and its linked promoter may partially be related to occupying a similar “chromatin domain,” which is relatively permissive of transcription. However, even though the cell-type specificity of the enhancer and promoter are clearly correlated (), transcription rates in general from the enhancer and promoter show virtually no correlation (Fig. S2B). In addition, it appears unlikely that enhancer transcription is simply a consequence of being linked to an active promoter, with the eRNA being produced by spurious RNAP II transcription as has been proposed previously, given the lack of correlation between promoters and enhancers. Collectively, our work indicates that eRNA production is specific to a selected group of cell-type specific enhancers.
By focusing on a single locus, an enhancer linked to Nanog, we confirmed that eRNA production at this genomic region was tightly linked to the pluripotent state. We found very similar results whether we primed our reverse transcription reaction with random hexamers or oligo-dT, implying that at least a fraction of eRNAs produced from this locus is polyadenylated to some extent. This remains an area of controversy, with one group reporting in neurons that eRNAs lack polyadenylation,Citation14 but other reports indicating that eRNAs can be polyadenylated.Citation13,Citation16 The RNA-seq data set used to identify eRNA production utilizes a genome-wide variant of nuclear run-off assays, and is therefore not affected by the presence or absence of polyadenylation for detection of eRNAs. Importantly, given that the polyadenylation of mRNAs tends to be rather long (typically >200 nts), the results from oligo dT priming may simply be related to the presence of a much shorter polyA tract (<20 nt), which could be related simply to the presence of a AT rich region, or perhaps a modified polyadenylation process, distinct from the classical pathway utilized in the cytoplasm during mRNA biogenesis. In addition, while the Nanog-linked enhancer is bidirectionally transcribed (), most reads arise from the minus strand. The work of others indicates that enhancers transcribed mostly unidirectionally typically produce polyadenylated transcripts,Citation16 consistent with our own findings. This issue remains difficult to resolve and, in fact, it may be a cell-type-dependent effect that requires substantial further study.
Perhaps the most surprising result was the clear segregation between ESC-specific eRNA-producing enhancers and eRNA-negative enhancers in terms of their Tet1 binding and DNA methylation profiles. Seminal work by Dirk Schübeler’s group indicates that distal regulatory elements exhibit either low (LMR) or unmethylated (UMR) regions, which are maintained by the binding of sequence specific factors.Citation39 In addition, two recent papers have shown that active DNA demethylation is critical to maintain enhancers in a hypomethylated state.Citation41,Citation50 Both groups observed an increase in oxidized cytosine species (5′ formylcytosine and/or 5′ carboxcytosine) after TDG depletion at enhancers. While there were subtle differences between the two papers, at least one groupCitation41 observed increased dependence on active DNA demethylation at poised (H3K4me1+/H3K27Ac−) vs. active (H3K4me1+/H3K27Ac+) enhancers, consistent with our observations of Tet1 occupancy (). This is an interesting distinction between the two, although our own reanalysis of 5mC levels in these enhancers show very little difference between the two ( and data not shown), implying that an alternative demethylation pathway may exist at active enhancers, perhaps mediated by activation induced deaminase (AID).Citation51 Recent work published while this manuscript was in preparation demonstrates the interaction between Tet proteins and the enzyme O-linked N-Acetylglucosamine Transferase (OgtCitation52-Citation54) raising the important question as to whether Ogt recruitment to enhancers may be critical to their eRNA production, perhaps through Ogt enzymatically altering the C-terminal domain (CTD) of RNAP II.Citation55 Importantly, the ability of Tet2 to bind OGT and permit histone O-GlcNAcylation was independent of Tet2 DNA hydroxylase activity.Citation52 Given recent work showing that Nanog can directly interact with Tet proteins and recruit them to specific loci,Citation56 it will be important to determine if this process is dependent upon DNA hydroxylation by Tet proteins, O-GlcNAcylation by Ogt, or a combination of the two to alter the epigenetic code of an enhancer to ensure proper eRNA production and function.
Recent work from the ENCODE project highlights that the genome contains at least an order of magnitude more enhancers than protein-coding loci.Citation3 Understanding which of these enhancer elements are critical to lineage-specific gene expression remains a daunting challenge. While distinctions between active and poised enhancers are well established, other, additional criteria would be useful, especially if they could be used to identify enhancers that are critical to lineage-specific transcriptional programs. Our own work, along with a recent description of super enhancers in ESCs,Citation28 identified specific genomic regions as containing pluripotent-critical enhancers, based upon different criteria, which are likely functionally equivalent. Thus, given that performing RNA-seq on tissues/cell-lines is technically more feasible than identifying genomic occupancy of multiple lineage-specific TFs, eRNA production may be an effective way to identify lineage-critical enhancers. Recent data derived from genome-wide chromosomal interaction studies indicate that eRNA producing enhancers form more interactions with other genomic elements than with transcriptionally silent enhancers,Citation12,Citation37 further emphasizing that eRNA production is an excellent marker to identify important cis-regulatory elements.
Materials and Methods
ChIP-Seq data set analysis
All data sets were directly downloaded (SRA format) from the GEO omnibus (Table S1) and reanalyzed as follows. Raw sequence files were aligned to mm9/NCBI Build 37 using Bowtie (−e 70, n 2, l 28, k 1).Citation57 Peaks were identified using a two-sided approach by MACS by comparing to a reference sample from the same experiment, typically input, sheared, un-precipitated genomic DNA.Citation58 We used a P value of <10−8 to identify high-confidence peaks for histones and 10−6 for non-histone binding events; to identify histone peaks with a relatively low false-negative rate we choose a 3 log higher P value (< 10−5). The individual files utilized, P values, other criteria used during identification, and original references are included in Table S1. All peak lists were compared with the peak lists from the original study (where available) to ensure there was at least a 50% concordance in the called peaks. To identify enhancers, a filtering procedure was used to eliminate other cis-acting elements. First, all peaks for the different groups (NOSM, H3K4me1, H3K27Ac, p300/CBP) that were within 2 kb upstream or downstream of the transcriptional start site (TSS) of a well-annotated gene or microRNA site were removed. Next, the remaining peaks were split into intragenic and extragenic peaks. Extragenic and intragenic peaks that overlapped with a region of H3K4me3 (P value of 10−5) were eliminated to remove unannotated promoters/genes or other classes of ncRNAs (such as lincRNAs). Extragenic peaks that overlapped with H3K36me3 (P value of 10−5) were eliminated for the same reason; this could not be used for intragenic peaks since many intronic and or exonic enhancers may show some degree of H3K36me3 enrichment. We also restricted H3K4me1 and H3K27Ac peaks to a size >1 kb. A list how many peaks were removed by each filtering step is provided in Table S5. For RNAP II, we identified regions occupied by RNAP II total or RNAP II Ser5p (the initiating form) that were not co-occupied by RNAP II Ser2p (the elongating form). To assign these enhancers to genes, we utilized a distance-based approach. Intragenic enhancers were assigned to the promoter within they resided. For extragenic enhancers, they were assigned to the TSS of the nearest gene within 50kb, either upstream or downstream. A list of enhancers, the genes they were linked to (if present), and the histone mark of the promoter are listed in Table S2. Super enhancersCitation28 were taken directly from the supplemental material provided with the manuscript.
Venn diagram generation
To generate the Venn Diagrams showing the common and unique regions in each set of enhancers we used pybedtools and bedtools. In particular, to be able to compare the # intersection in each subset of the Venn diagram corresponding to different classes we used a symmetric intersection operation. This means that given two sets of regions A and B, the order of the intersection operation ∩ does not change the number of intersections i.e., # (A ∩ B) = # (B ∩ A). This is especially useful when a coordinate in a given set span several coordinates in another set, in this case we count always one intersection.
RNA-seq analysis
Aligned reads GRO-seq analysis was downloaded from the GEO omnibus (GSE27037). For extragenic enhancers, RNA reads from both strands were counted using the BEDTools suite. For intragenic enhancers to prevent counting reads from sense-strand gene transcription, only the anti-sense strand was counted. RPKMs were assigned by normalizing to the total number of reads from an experiment and the size (in kb) of the genomic feature.
Scatter Plot
The scatter plot reports the Pearson's correlation between the RNA levels of enhancer-gene pairs; each pair was obtained mapping the enhancer to its closest gene. The enhancer eRNA level (y-axis) and the gene mRNA level (x-axis) are quantified in RPKM. The RPKM range 0–2 in both axes was divided into 30 bins, obtaining 900 tiles in the xy plane. The color of each tile is proportional to the number of points presented in it.
Tet1 enrichment
Chip-seq data sets for Tet1 was downloaded from the GEO omnibus (GSE26832). The aligned files for Tet1 and Tet1 mock were processed by counting the reads genome-wide in non-overlapping windows of 200 bp and reporting the normalized reads per million (RPM). The ratio between Tet1 and Tet1 mock in RPM was computed and the Tet1 average profiles and heatmaps were built using a 5 kb window at each peak center of the different classes of enhancers.
DNA methylation
Bisulfite-Seq data from GEO Omnibus (GSE30202) was downloaded to obtain the methylation levels in the different classes of enhancers. In particular, the coverage and methylated counts for all CpG regions were used. The average methylation level in a given region was calculated as the ratio between the numbers of reads mapped to any CpG and the sum of the methylated counts inside the region. A track with a resolution of 50 bp was used to build heatmaps and the average profiles for the different enhancers.
RA differentiation data set and Gene Set Enrichment Analysis (GSEA) analysis
A time course of retinoic acid differentiation on ESC was downloaded (GSE4679), RMA normalized, and a comparison made between the Day 0 and Day 6 samples of retinoic acid differentiation. GSEA was utilized and normalized enrichment scores determined for the genes linked to different enhancers. Only gene sets with P < 0.05 and False Discovery Rate (FDR) < 10% were considered statistically significant.
Protein, RNA isolation and Quantitative PCR
Total RNA was harvested from cells following manufacturer’s protocol (Trizol, Invitrogen). Genomic DNA was removed from total RNA samples using a DNase digestion step and passing RNA over a column following manufacturer’s protocol (RNeasy Micro, Qiagen). Equal amounts of DNA-free total RNA were converted to cDNA using either the iScript cDNA synthesis kit (Biorad), or for specific priming (random hexamers or Oligo dT) using the Superscript III kit (Invitrogen). Primers used are listed in Table S4 and quantitative PCR (qPCR) was performed on a BioRad iCycler. Quantifications were normalized to an internal control (Actin for RT-qPCR and GAPDH for ChIP-qPCR).Citation25 Whole cell extracts were prepared as before,Citation25 total protein measured by a modified Bradford Assay (BioRad), and equal amounts of total protein separated by SDS-PAGE electrophoresis using standard techniques. Antibodies used for western blotting and/or ChIP: anti-Sall4 (Abcam, ab29112), anti-Nanog (Millipore, AB5731), anti-Tet1 (Millipore, 09-872), anti-Oct4 (Abcam, ab19857), anti GAPDH-HRP (Santa Cruz, sc-25778), donkey anti-rabbit IgG-HRP (Santa Cruz, sc-2609). Anti-RNAP II (Covance, 8WG16), anti-RPC32 (Santa Crux, sc-21754), and anti-Tet2 (generous gift of Dr Xiaochun Yu, University of MichiganCitation52).
Cell Culture, RA differentiation
Gelatin-Adapted CJ9 cells were utilized for all experiments listed. These are a 129svj derived murine ESC line similar to the one we have used previously and were cultured under similar conditions.Citation25
Chromatin Immunoprecipitation (ChIP)
Antibody-mediated chip was modified from the method described by.Citation59 Cells were cross-linked with 1% formaldehyde for 5 min, and quenched with 125 mM Glycine, 5 min rocking at room temperature. Cells were washed with PBS −/−, collected, and resuspended in 1ml IP Buffer (150 mM NaCl, 50 mM Tris-HCl, 5 mM EDTA, 1% TritonX-100, 0.5% NP-40) plus protease inhibitors (1:1000 Protease Inhibitors cocktail (Sigma, P8340-5ML) and 5:1000 PMSF (Sigma, 93482-50ML-F), transferred to 2ml microtubes, and nuclei were pelleted by centrifugation at max speed for one minute at 4 °C. The nuclei were resuspended in 1ml IP Buffer plus inhibitors and sheared by sonication with a Misonix 3000 and microtip to an approximate size of 200–500 bp. The insoluble fraction was pelleted by centrifugation, and the appropriate antibodies added (typically 1–2 μg/500 μL of sheared chromatin) to the supernatant and incubated with rotation overnight at 4 °C. The next day pre-washed Protein A Dynabeads (Invitrogen) were added to the antibody:chromatin and mixed for 90 min at 4 °C. Beads were then bound to a magnet column and washed once with IP buffer (without protease inhibitors), once with High Salt Buffer (500 mM NaCl, 50 mM HEPES, 1mM EDTA, 1% TritonX-100, 0.1% Deoxycholate), once with LiCl Buffer (250 mM LiCl, 10 mM Tris, 1 mM EDTA, 0.5% NP40, 0.5% Deoxycholate) and once TE Buffer (10 mM Tris, 1 mM EDTA). After final wash the beads were resuspended in 150 μl SDS Elution Buffer (1% SDS, 50 mM Tris, 10 mM EDTA), and reverse cross-linked/eluted overnight at 65 °C. The supernatant was isolated using a magnet the next day. Reverse cross-linked chromatin was treated with RNase A, Proteinase K, and then phenol:chloroform extracted and precipitated overnight with ethanol and sodium acetate. DNA was quantified by fluorometric quantitation (Qubit, Invitrogen), and equal amount of chromatin were used for downstream applications. Detailed protocol available upon request.
Metabolically labeled cell lines that express biotinylatable versions of NanogCitation43 and Sall4a/Sall4bCitation25 have been described previously. Biotin-mediated ChIP (bioChIP) was performed similar to our previous publication.Citation25 All ChIPs were quantified using real-time PCR on a BioRad iCycler using SYBR Green (Biorad). All ChIP data are normalized to sheared, non-immunoprecipitated genomic DNA.
Lentiviral generation and infections
Lentiviral constructs developed by the RNAi Consortium (TRC) were obtained from either Open Biosystems or Sigma-Aldrich. The following constructs were utilized for each mRNA: pLKO.1 is the parental viral vector without shRNA. Sall4 (TRCN0000097821, TRCN0000097824), Nanog (TRCN0000075336, TRCN0000075337), Tet1 (shRNA #1: TRCN0000341848, shRNA#2: TRCN0000341849), and Tet2 (shRNA #1:TRCN0000250893, shRNA #2 TRCN0000250895). Lentiviruses were generated by transiently transfecting 293T packaging cells with the viral vector, VSV-G, and the packaging plasmid pΔ8.9 using Polyethylenimine (PEI) based method similar to (ref. Citation35; detailed protocol available upon request). Virus was harvested at 48 and 72 h post infection and directly applied to ESCs cells plated the day before after being supplemented with polybrene (final concentration of 10 μg/mL). After a total of two rounds of infection (two days), the viral supernatants were removed from the CJ9 cells and ESC media supplemented with puromycin (final concentration of 4 μg/mL) was applied to the cells for a total of two days to select for cells with a high multiplicity of infection. Cells were then harvested for either RNA or protein extraction and treated as detailed above.
Reporter plasmid generation
The Nanog promoter (chr6:122 656 172–122 657 706; 1535 bp, mm9) and enhancer (chr6:122 611 603–122 614 453; 2850 bp, mm9) were amplified directly from a BAC (RP2-19O18). Both regions were fully sequenced and matched the genomic reference sequence. The Nanog promoter was cloned into the KpnI/Xho I sites of the firefly luciferase vector pGL2-basic (Promega). The enhancer was subsequently cloned into the SalI site of this plasmid, i.e., downstream of the luciferase coding region, or into the same site of the pGL2-promoter vector (Promega), which contains a SV40 derived minimal promoter. In both cases, both orientations were cloned. Plus orientation indicates that the plus strand of the genomic region is on the same strand as the coding luciferase sequence.
Transient Transfection Assay
The above plasmids were transfected transiently into either CJ9 cells or NIH-3T3 cells using Lipofectamine 2000 (Invitrogen) along with a control plasmid (Renilla Luciferase, pRL-EF). Thirty-six hours after transfection cells were lysed and luciferase activity measured using the Dual-Luciferase Reporter Assay kit (Promega) following the manufacturer’s protocol. Luminescence was measured on a Wallac Victor V1420 (PerkinElmer). Background levels of luminescence were obtained from untransfected control cells treated in parallel and subtracted from the luminescence of experimental samples. Firefly luciferase activity was normalized to Renilla luciferase activity following manufacturer’s protocol.
Bisulfite sequencing
Cells were infected with lentiviruses encoding the empty virus (pLKO.1), or shRNAs #1 to Sall4, Nanog, or Tet1 as above, and after two days of puromycin treatment genomic DNA was isolated using the Qiagen DNeasy kit following manufacturer’s protocol). Bisulfite conversion and cleanup were performed using the EpiTect bisulfite kit (Qiagen). Nested PCRs were performed using the primer pairs indicated in Table S4—primer sequences were developed utilizing MethPrimer (http://www.urogene.org/methprimer/). For the first (outer) round of PCR, the following amplification conditions were used: 95 °C × 5min, 95 °C × 30 s, 54 °C × 30 s, 72 °C × 3 min for five cycles, then 95 °C × 30 s, 54 °C × 30 s, 72 °C × 45 s for 25 additional cycles, 72 °C × 10 min. A small aliquot of the first (outer) PCR was used as a template for the second (inner) PCR, with the following amplification conditions 95 °C × 5min, 95 °C × 30 s, 54 °C × 30 s, 72 °C × 30 s for 25–35 cycles total, 72 °C × 10 min. PCR products were gel purified and cloned using the TOPO-TA cloning method (Invitrogen). Twelve minipreps were prepared for each condition and sequenced (Retrogen). Bisulfite data was analyzed and graphs prepared using QUMA (http://quma.cdb.riken.jp)
| Abbreviations: | ||
| eRNA | = | enhancer-transcribed RNA |
| ESC | = | embryonic stem cell |
| CBP | = | CREB-binding protein |
| TF | = | transcription factor |
| DNase HS | = | DNase hypersensitivity site |
| RNAP II | = | RNA polymerase II |
| ncRNA | = | non-coding RNA |
| lincRNAs | = | Long intergenic non-coding RNA |
| CTCF | = | CCCTC-binding factor |
| NOS | = | genomic regions occupied by Nanog, Oct4, Sox2 |
| NOSM | = | genomic regions occupied by Nanog, Oct4, Sox2, and either Mediator1 or Mediator12. |
| LMR | = | low methylated region |
| UMR | = | unmethylated region |
| FMR | = | fully methylated region |
| GRO-seq | = | genome wide nuclear run-off assay coupled with next generation sequencing. |
| MEFs | = | mouse embryonic fibroblast |
| RA | = | retinoic acid |
| RPKM | = | reads per kilobase per million reads |
| NP | = | neural progenitors |
| ChIP | = | chromatin immunoprecipitation |
| qPCR | = | quantitative PCR |
| bioChIP | = | biotin-tagged chromatin immunoprecipitation |
| Ser2p | = | RNAP II phosphorylated on Serine 2 |
| Ser5p | = | RNAP II phosphorylated on Serine 5 |
| GSEA | = | gene set enrichment analysis |
| RT | = | reverse transcriptase |
| HMM | = | hidden Markov model |
| AID | = | activation deaminase |
| Ogt | = | O-linked N-acetylglucosamine transferase |
Additional material
Download Zip (6.4 MB)Acknowledgments
The authors would like to thank all the groups who placed their various datasets into the NCBI database, many of whom provided us with additional information and/or guidance with regards to analysis of their respective datasets. We would like to thank Dr Xiaochun Xu (Univ. of Michigan) for generously providing the anti-Tet2 antibody used in this work. This work is funded in part by NHLBI (HL087951 to Rao S) and NHGRI (HG005085 to Yuan G). In addition, Rao S is supported by a pilot grant from the American Cancer Society (Institutional Research Grant #86-004) and the Midwest Athletes against Childhood Cancer.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Rao S.. Embryonic Stem Cells: A Perfect Tool for Studying Mammalian Transcriptional Enhancers. J Stem Cell Res Ther 2012; S10:007; http://dx.doi.org/10.4172/2157-7633.S10-007
- Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, Sheffield NC, Stergachis AB, Wang H, Vernot B, et al. The accessible chromatin landscape of the human genome. Nature 2012; 489:75 - 82; http://dx.doi.org/10.1038/nature11232; PMID: 22955617
- ENCODE Project Consortium. Bernstein BE, Birney E, Dunham I, Green ED, Gunter C, Snyder M. Consortim. An integrated encyclopedia of DNA elements in the human genome. Nature 2013; 488:57 - 74; PMID: 22955616
- Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet 2007; 39:311 - 8; http://dx.doi.org/10.1038/ng1966; PMID: 17277777
- Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW, et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 2009; 459:108 - 12; http://dx.doi.org/10.1038/nature07829; PMID: 19295514
- Visel A, Blow MJ, Li Z, Zhang T, Akiyama JA, Holt A, Plajzer-Frick I, Shoukry M, Wright C, Chen F, et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature 2009; 457:854 - 8; http://dx.doi.org/10.1038/nature07730; PMID: 19212405
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A 2010; 107:21931 - 6; http://dx.doi.org/10.1073/pnas.1016071107; PMID: 21106759
- Zentner GE, Tesar PJ, Scacheri PC. Epigenetic signatures distinguish multiple classes of enhancers with distinct cellular functions. Genome Res 2011; 21:1273 - 83; http://dx.doi.org/10.1101/gr.122382.111; PMID: 21632746
- Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 2011; 470:279 - 83; http://dx.doi.org/10.1038/nature09692; PMID: 21160473
- Whyte WA, Bilodeau S, Orlando DA, Hoke HA, Frampton GM, Foster CT, Cowley SM, Young RA. Enhancer decommissioning by LSD1 during embryonic stem cell differentiation. Nature 2012; 482:221 - 5; PMID: 22297846
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012; 485:376 - 80; http://dx.doi.org/10.1038/nature11082; PMID: 22495300
- Sanyal A, Lajoie BR, Jain G, Dekker J. The long-range interaction landscape of gene promoters. Nature 2012; 489:109 - 13; http://dx.doi.org/10.1038/nature11279; PMID: 22955621
- Wang D, Garcia-Bassets I, Benner C, Li W, Su X, Zhou Y, Qiu J, Liu W, Kaikkonen MU, Ohgi KA, et al. Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature 2011; 474:390 - 4; http://dx.doi.org/10.1038/nature10006; PMID: 21572438
- Kim T-K, Hemberg M, Gray JM, Costa AM, Bear DM, Wu J, Harmin DA, Laptewicz M, Barbara-Haley K, Kuersten S, et al. Widespread transcription at neuronal activity-regulated enhancers. Nature 2010; 465:182 - 7; http://dx.doi.org/10.1038/nature09033; PMID: 20393465
- De Santa F, Barozzi I, Mietton F, Ghisletti S, Polletti S, Tusi BK, Muller H, Ragoussis J, Wei C-L, Natoli G. A large fraction of extragenic RNA pol II transcription sites overlap enhancers. PLoS Biol 2010; 8:e1000384; http://dx.doi.org/10.1371/journal.pbio.1000384; PMID: 20485488
- Koch F, Fenouil R, Gut M, Cauchy P, Albert TK, Zacarias-Cabeza J, Spicuglia S, de la Chapelle AL, Heidemann M, Hintermair C, et al. Transcription initiation platforms and GTF recruitment at tissue-specific enhancers and promoters. Nat Struct Mol Biol 2011; 18:956 - 63; http://dx.doi.org/10.1038/nsmb.2085; PMID: 21765417
- Natoli G, Andrau J-C. Noncoding transcription at enhancers: general principles and functional models. Annu Rev Genet 2012; 46:1 - 19; http://dx.doi.org/10.1146/annurev-genet-110711-155459; PMID: 22905871
- Melo CA, Drost J, Wijchers PJ, van de Werken H, de Wit E, Oude Vrielink JA, Elkon R, Melo SA, Léveillé N, Kalluri R, et al. eRNAs are required for p53-dependent enhancer activity and gene transcription. Mol Cell 2013; 49:524 - 35; http://dx.doi.org/10.1016/j.molcel.2012.11.021; PMID: 23273978
- Li W, Notani D, Ma Q, Tanasa B, Nunez E, Chen AY, Merkurjev D, Zhang J, Ohgi K, Song X, et al. Functional roles of enhancer RNAs for oestrogen-dependent transcriptional activation. Nature 2013; 498:516 - 20; http://dx.doi.org/10.1038/nature12210; PMID: 23728302
- Lam MTY, Cho H, Lesch HP, Gosselin D, Heinz S, Tanaka-Oishi Y, Benner C, Kaikkonen MU, Kim AS, Kosaka M, et al. Rev-Erbs repress macrophage gene expression by inhibiting enhancer-directed transcription. Nature 2013; 498:511 - 5; http://dx.doi.org/10.1038/nature12209; PMID: 23728303
- Guttman M, Donaghey J, Carey BW, Garber M, Grenier JK, Munson G, Young G, Lucas AB, Ach R, Bruhn L, et al. lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature 2011; 477:295 - 300; http://dx.doi.org/10.1038/nature10398; PMID: 21874018
- Cabili MN, Trapnell C, Goff L, Koziol M, Tazon-Vega B, Regev A, Rinn JL. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev 2011; 25:1915 - 27; http://dx.doi.org/10.1101/gad.17446611; PMID: 21890647
- Orkin SH, Hochedlinger K. Chromatin connections to pluripotency and cellular reprogramming. Cell 2011; 145:835 - 50; http://dx.doi.org/10.1016/j.cell.2011.05.019; PMID: 21663790
- Young RA. Control of the embryonic stem cell state. Cell 2011; 144:940 - 54; http://dx.doi.org/10.1016/j.cell.2011.01.032; PMID: 21414485
- Rao S, Zhen S, Roumiantsev S, McDonald LT, Yuan GC, Orkin SH. Differential roles of Sall4 isoforms in embryonic stem cell pluripotency. Mol Cell Biol 2010; 30:5364 - 80; http://dx.doi.org/10.1128/MCB.00419-10; PMID: 20837710
- Jaenisch R, Young R. Stem cells, the molecular circuitry of pluripotency and nuclear reprogramming. Cell 2008; 132:567 - 82; http://dx.doi.org/10.1016/j.cell.2008.01.015; PMID: 18295576
- Ivanova N, Dobrin R, Lu R, Kotenko I, Levorse J, DeCoste C, Schafer X, Lun Y, Lemischka IR. Dissecting self-renewal in stem cells with RNA interference. Nature 2006; 442:533 - 8; http://dx.doi.org/10.1038/nature04915; PMID: 16767105
- Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, Young RA. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 2013; 153:307 - 19; http://dx.doi.org/10.1016/j.cell.2013.03.035; PMID: 23582322
- Pekowska A, Benoukraf T, Zacarias-Cabeza J, Belhocine M, Koch F, Holota HELEN, Imbert J, Andrau J-C, Ferrier P, Spicuglia S. H3K4 tri-methylation provides an epigenetic signature of active enhancers. EMBO J 2011; 30:4198 - 210; http://dx.doi.org/10.1038/emboj.2011.295; PMID: 21847099
- Seila AC, Calabrese JM, Levine SS, Yeo GW, Rahl PB, Flynn RA, Young RA, Sharp PA. Divergent transcription from active promoters. Science 2008; 322:1849 - 51; http://dx.doi.org/10.1126/science.1162253; PMID: 19056940
- Min IM, Waterfall JJ, Core LJ, Munroe RJ, Schimenti J, Lis JT. Regulating RNA polymerase pausing and transcription elongation in embryonic stem cells. Genes Dev 2011; 25:742 - 54; http://dx.doi.org/10.1101/gad.2005511; PMID: 21460038
- Rahl PB, Lin CY, Seila AC, Flynn RA, McCuine S, Burge CB, Sharp PA, Young RA. c-Myc regulates transcriptional pause release. Cell 2010; 141:432 - 45; http://dx.doi.org/10.1016/j.cell.2010.03.030; PMID: 20434984
- Kim J, Woo AJ, Chu J, Snow JW, Fujiwara Y, Kim CG, Cantor AB, Orkin SH. A Myc network accounts for similarities between embryonic stem and cancer cell transcription programs. Cell 2010; 143:313 - 24; http://dx.doi.org/10.1016/j.cell.2010.09.010; PMID: 20946988
- Wang KC, Yang YW, Liu B, Sanyal A, Corces-Zimmerman R, Chen Y, Lajoie BR, Protacio A, Flynn RA, Gupta RA, et al. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature 2011; 472:120 - 4; http://dx.doi.org/10.1038/nature09819; PMID: 21423168
- Toledo JR, Prieto Y, Oramas N, Sánchez O. Polyethylenimine-based transfection method as a simple and effective way to produce recombinant lentiviral vectors. Appl Biochem Biotechnol 2009; 157:538 - 44; http://dx.doi.org/10.1007/s12010-008-8381-2; PMID: 19089654
- Shen Y, Yue F, McCleary DF, Ye Z, Edsall L, Kuan S, Wagner U, Dixon J, Lee L, Lobanenkov VV, et al. A map of the cis-regulatory sequences in the mouse genome. Nature 2012; 488:116 - 20; http://dx.doi.org/10.1038/nature11243; PMID: 22763441
- Phillips-Cremins JE, Sauria MEG, Sanyal A, Gerasimova TI, Lajoie BR, Bell JSK, Ong C-T, Hookway TA, Guo C, Sun Y, et al. Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell 2013; 153:1281 - 95; http://dx.doi.org/10.1016/j.cell.2013.04.053; PMID: 23706625
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005; 102:15545 - 50; http://dx.doi.org/10.1073/pnas.0506580102; PMID: 16199517
- Stadler MB, Murr R, Burger L, Ivanek R, Lienert F, Schöler A, van Nimwegen E, Wirbelauer C, Oakeley EJ, Gaidatzis D, et al. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature 2011; 480:490 - 5; PMID: 22170606
- Wu H, D’Alessio AC, Ito S, Xia K, Wang Z, Cui K, Zhao K, Sun YE, Zhang Y. Dual functions of Tet1 in transcriptional regulation in mouse embryonic stem cells. Nature 2011; 473:389 - 93; http://dx.doi.org/10.1038/nature09934; PMID: 21451524
- Shen L, Wu H, Diep D, Yamaguchi S, D’Alessio AC, Fung H-L, Zhang K, Zhang Y. Genome-wide analysis reveals TET- and TDG-dependent 5-methylcytosine oxidation dynamics. Cell 2013; 153:692 - 706; http://dx.doi.org/10.1016/j.cell.2013.04.002; PMID: 23602152
- Levasseur DN, Wang J, Dorschner MO, Stamatoyannopoulos JA, Orkin SH. Oct4 dependence of chromatin structure within the extended Nanog locus in ES cells. Genes Dev 2008; 22:575 - 80; http://dx.doi.org/10.1101/gad.1606308; PMID: 18283123
- Wang J, Rao S, Chu J, Shen X, Levasseur DN, Theunissen TW, Orkin SH. A protein interaction network for pluripotency of embryonic stem cells. Nature 2006; 444:364 - 8; http://dx.doi.org/10.1038/nature05284; PMID: 17093407
- Wu Q, Chen X, Zhang J, Loh YH, Low TY, Zhang W, Zhang W, Sze SK, Lim B, Ng HH. Sall4 interacts with Nanog and co-occupies Nanog genomic sites in embryonic stem cells. J Biol Chem 2006; 281:24090 - 4; http://dx.doi.org/10.1074/jbc.C600122200; PMID: 16840789
- Das S, Jena S, Levasseur DN. Alternative splicing produces Nanog protein variants with different capacities for self-renewal and pluripotency in embryonic stem cells. J Biol Chem 2011; 286:42690 - 703; http://dx.doi.org/10.1074/jbc.M111.290189; PMID: 21969378
- Zhang J, Tam W-L, Tong GQ, Wu Q, Chan H-Y, Soh B-S, Lou Y, Yang J, Ma Y, Chai L, et al. Sall4 modulates embryonic stem cell pluripotency and early embryonic development by the transcriptional regulation of Pou5f1. Nat Cell Biol 2006; 8:1114 - 23; http://dx.doi.org/10.1038/ncb1481; PMID: 16980957
- Ito S, D’Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 2010; 466:1129 - 33; http://dx.doi.org/10.1038/nature09303; PMID: 20639862
- Koh KP, Yabuuchi A, Rao S, Huang Y, Cunniff K, Nardone J, Laiho A, Tahiliani M, Sommer CA, Mostoslavsky G, et al. Tet1 and Tet2 regulate 5-hydroxymethylcytosine production and cell lineage specification in mouse embryonic stem cells. Cell Stem Cell 2011; 8:200 - 13; http://dx.doi.org/10.1016/j.stem.2011.01.008; PMID: 21295276
- Williams K, Christensen J, Pedersen MT, Johansen JV, Cloos PAC, Rappsilber J, Helin K. TET1 and hydroxymethylcytosine in transcription and DNA methylation fidelity. Nature 2011; 473:343 - 8; http://dx.doi.org/10.1038/nature10066; PMID: 21490601
- Song C-X, Szulwach KE, Dai Q, Fu Y, Mao S-Q, Lin L, Street C, Li Y, Poidevin M, Wu H, et al. Genome-wide profiling of 5-formylcytosine reveals its roles in epigenetic priming. Cell 2013; 153:678 - 91; http://dx.doi.org/10.1016/j.cell.2013.04.001; PMID: 23602153
- Cortellino S, Xu J, Sannai M, Moore R, Caretti E, Cigliano A, Le Coz M, Devarajan K, Wessels A, Soprano D, et al. Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell 2011; 146:67 - 79; http://dx.doi.org/10.1016/j.cell.2011.06.020; PMID: 21722948
- Chen Q, Chen Y, Bian C, Fujiki R, Yu X. TET2 promotes histone O-GlcNAcylation during gene transcription. Nature 2013; 493:561 - 4; http://dx.doi.org/10.1038/nature11742; PMID: 23222540
- Vella P, Scelfo A, Jammula S, Chiacchiera F, Williams K, Cuomo A, Roberto A, Christensen J, Bonaldi T, Helin K, et al. Tet proteins connect the O-linked N-acetylglucosamine transferase Ogt to chromatin in embryonic stem cells. Mol Cell 2013; 49:645 - 56; http://dx.doi.org/10.1016/j.molcel.2012.12.019; PMID: 23352454
- Deplus R, Delatte B, Schwinn MK, Defrance M, Méndez J, Murphy N, Dawson MA, Volkmar M, Putmans P, Calonne E, et al. TET2 and TET3 regulate GlcNAcylation and H3K4 methylation through OGT and SET1/COMPASS. EMBO J 2013; 32:645 - 55; http://dx.doi.org/10.1038/emboj.2012.357; PMID: 23353889
- Ranuncolo SM, Ghosh S, Hanover JA, Hart GW, Lewis BA. Evidence of the involvement of O-GlcNAc-modified human RNA polymerase II CTD in transcription in vitro and in vivo. J Biol Chem 2012; 287:23549 - 61; http://dx.doi.org/10.1074/jbc.M111.330910; PMID: 22605332
- Costa Y, Ding J, Theunissen TW, Faiola F, Hore TA, Shliaha PV, Fidalgo M, Saunders A, Lawrence M, Dietmann S, et al. NANOG-dependent function of TET1 and TET2 in establishment of pluripotency. Nature 2013; 495:370 - 4; http://dx.doi.org/10.1038/nature11925; PMID: 23395962
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 2009; 10:R25; http://dx.doi.org/10.1186/gb-2009-10-3-r25; PMID: 19261174
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol 2008; 9:R137; http://dx.doi.org/10.1186/gb-2008-9-9-r137; PMID: 18798982
- Nelson JD, Denisenko O, Bomsztyk K. Protocol for the fast chromatin immunoprecipitation (ChIP) method. Nat Protoc 2006; 1:179 - 85; http://dx.doi.org/10.1038/nprot.2006.27; PMID: 17406230