
Abstract
The intrauterine environment has the potential to “program” the developing fetus in a way that can be potentially deleterious to later health. While in utero environmental/stochastic factors are known to influence DNA methylation profile at birth, it has been difficult to assign specific examples of epigenetic variation to specific environmental exposures. Recently, several studies have linked exposure to smoking with DNA methylation change in the aryl hydrocarbon receptor repressor (AHRR) gene in blood. This includes hypomethylation of AHRR in neonatal blood in response to maternal smoking in pregnancy. The role of AHRR as a negative regulator of pathways involved in pleiotropic responses to environmental contaminants raises the possibility that smoking-induced hypomethylation is an adaptive response to an adverse in utero environmental exposure. However, the tissue specificity of the response to maternal smoking, and the stability of the methylation changes early in life remain to be determined. In this study we analyzed AHRR methylation in three cell types—cord blood mononuclear cells (CBMCs), buccal epithelium, and placenta tissue—from newborn twins of mothers who smoked throughout pregnancy and matched controls. Further, we explored the postnatal stability of this change at 18 months. Our results confirm the previous association between maternal smoking and AHRR methylation in neonatal blood. In addition, this study expands the region of AHRR methylation altered in response to maternal smoking during pregnancy and reveals the tissue-specific nature of epigenetic responses to environmental exposures in utero. Further, the evidence for postnatal stability of smoking-induced epigenetic change supports a role for epigenetics as a mediator of long-term effects of specific in utero exposures in humans. Longitudinal analysis of further specific exposures in larger cohorts is required to examine the extent of this phenomenon in humans.
Introduction
During pregnancy the developing fetus must adapt to its environment in order to optimize growth and to minimize the potential adverse effects of harmful environmental exposures. While beneficial in the womb, such adaptations can also be potentially deleterious to the long-term health of an individual. For example, a fetus in a low nutrient environment may optimize its metabolic status in “anticipation” of a low caloric postnatal world, which would mismatch with a high nutrient postnatal diet. This fetal programmingCitation1 is postulated to explain the higher rates of obesity and diabetes in individuals born small for gestational age.Citation2,Citation3 The more widely applicable Developmental Origins of Health and Disease (DOHaD) hypothesis states that the intrauterine environment can “program” the fetus through subtle changes in organ structure or function, so as to predispose to disease in adulthood.Citation4,Citation5 Mounting evidence suggests a key role for epigenetic mechanisms (such as DNA methylation) in mediating this process.Citation6
Despite increasing association studies linking DNA methylation change to disease,Citation7,Citation8 little consistent and reproducible data has emerged linking specific environmental exposures to specific epigenetic change, a missing piece in the hypothesized DOHaD pathway. Epigenome Wide Association Studies (EWAS) have the potential to identify such changes,Citation9 and one of the largest EWAS performed to date involved the screening of 1062 newborn cord blood samples using the Infinium HumanMethylation450 platform (HM450) in an attempt to identify DNA methylation change in newborns associated with maternal smoking during pregnancy.Citation10 Methylation changes at four genes were identified: hypomethylation of the aryl hydrocarbon receptor repressor (AHRR) and growth factor independent 1 transcription repressor (GFI1), and hypermethylation of cytochrome P450–1A1 (CYP1A1) and myosin IG (MYO1G). AHRR is involved in the detoxification of chemicals found in tobacco smoke, and lower methylation may be a cellular response to the presence of these chemicals, resulting in higher expression of this gene. Interestingly, other recent studies in adults have also identified hypomethylation in the same region of the AHRR gene in association with smoking in lymphoblasts and alveolar macrophages,Citation11 whole blood,Citation12 and lymphocytes.Citation13 Further, a large study of more than 2000 adults, including 498 smokers, identified hypomethylation of this gene in whole blood of smokers.Citation14 Thus, five independent studies have now linked a decrease in AHRR methylation to smoking exposure with an effect size of between 0.075– 0.24 (~7.5–24% methylation).
Despite these findings, the full domain of methylation change across the AHRR gene in response to maternal smoking and the extent across different tissues remains to be determined. Similarly, the postnatal stability of the altered DNA methylation profile remains unclear. The aim of this study was to (1) replicate previous findings of an effect of maternal smoking on AHRR methylation in particular, as well as GFI1 and MYO1G methylation, (2) to expand the region of the gene assayed for methylation change, (3) to assess methylation change across multiple tissues and, (4) to assess postnatal stability of any methylation difference in the first years of life. Through the use of twin samples collected as part of the Peri/Postnatal Epigenetics Twin Study,Citation15,Citation16 we also sought to (5) explore the association between the underlying genetics and methylation at this locus.
Results
Characterization of tissue-specific DNA methylation patterns within the AHRR gene body
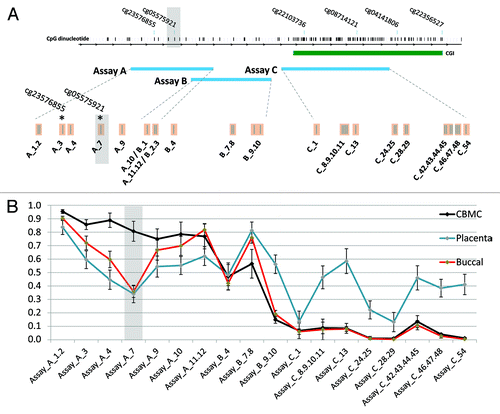
DNA methylation within intron 3 of the AHRR gene was measured using three overlapping assays covering 32 individual CpG sites contained in 18 measurable CpG units (; Fig. S1). This region contains the previously identified smoking-associated CpG site (HM450 probe cg05575921). DNA methylation was measured in three tissues from newborn twins: CBMCs (n = 46 pregnancies), buccal epithelium (n = 15 pregnancies), and placenta (n = 24 pregnancies). CBMCs and buccal epithelium showed intermediate to high methylation within the CpG island shore (flanking the CpG island), with an almost completely unmethylated pattern within the CpG island (CGI) (). Placenta on the other hand showed an intermediate methylation pattern throughout the CpG island shore and the island itself. This supports a previous finding that a CpG island within AHRR is monoallelically methylated in human first and third trimester placenta.Citation17 Of particular interest is the lower methylation at CpG_A7 (cg05575921) in buccal epithelium and placenta (average ~35% methylation) compared with CBMCs, which showed consistently high methylation at this site (average ~80% methylation) (). Coupled with the previously demonstrated enrichment for active histone marks at this region (Fig. S1A), this tissue specific methylation pattern supports a functional role for this region of the AHRR gene.
Figure 1. Tissue specific DNA methylation patterns within AHRR. (A) Map of the EpiTYPER assays (blue rectangles) covering the AHRR region of interest, showing analyzable CpG sites. Assay A and B cover the CpG site of interest (CpG_A7 - cg05575921) and surrounding CpG sites. The CGI is represented as a green rectangle. (B) DNA methylation level in CBMCs, buccal epithelium, and placenta show tissue-specific differences at CpG_A7. Grey shaded box = “cg05575921” CpG site of interest.
Maternal smoking throughout pregnancy is associated with AHRR hypomethylation specifically in cord blood mononuclear cells
Methylation data were separated into three groups for analysis: “smoked throughout,” “smoked early,” and “never smoked.” The “smoked throughout” group reported smoking prior to pregnancy, at the time they found out about the pregnancy and at each trimester (12, 24, and 36 wk). The “smoked early” group reported smoking up to the time of finding out about the pregnancy, but not thereafter (). Plasma cotinine levels were measured in a selection of maternal and infant samples and a strong correlation with questionnaire data was obtained at both time points (Table S2). The average level of methylation at CpG_A7 (corresponding to cg05575921) in CBMCs was 0.10 (10%) lower in the “smoked throughout” (0.73) compared with the “never smoked” (0.83) group (; Table S3), while the corresponding range of methylation within the groups was 0.62–0.89 and 0.71–0.91, respectively. Across the region assayed, 6 contiguous CpG analytical units (spanning 8 CpG sites) over 275 bp showed lower methylation in the smoking group (delta β > 0.05, P < 0.05; ). Interestingly, there were no differences in mean methylation between the “never smoked” and “smoked early” groups across assay A and B () and no difference in methylation at the CpG island (assay C), which was hypomethylated in all groups (). Of particular interest, we found no evidence for an association between maternal smoking and methylation at this region in buccal epithelium or the placenta of the same pregnancies (Fig. S2). However, it should be noted that due to lack of available tissue, buccal, and placenta analysis was limited to 7 and 8 smoking exposed pregnancies, respectively, and thus our study would have been unpowered to detect smaller methylation differences (~<6%) between smokers and non-smokers. Furthermore, there was no difference in methylation level between CBMCs from males and females (Fig. S3). These findings suggest that the effect of maternal smoking on AHRR methylation is tissue and timing specific, possibly requiring prolonged exposure in utero. However, in contrast to the widely held view that early pregnancy is an especially sensitive time point for environmentally induced epigenetic change, we found no evidence that first trimester smoking exposure in isolation has any effect on AHRR methylation levels in progeny.
Table 1. Summary of the study population
Figure 2. Association between maternal smoking and AHRR methylation in CBMCs. Maternal smoking throughout pregnancy is associated with lower methylation at (A) CpG_A7, (B) across assay A and (C) across assay B. Maternal smoking early in pregnancy was not associated with lower AHRR methylation. (D) 8 CpG sites, contained within 6 CpG units, show lower methylation (dB > 0.05, P < 0.05) in response to maternal smoking. The difference in methylation is limited to the CpG Island shore, whereas the CGI is hypomethylated in all CBMC samples. Y-axis = DNA methylation level. Grey shaded box = “cg05575921” CpG site of interest. Error bars = 95% CI.
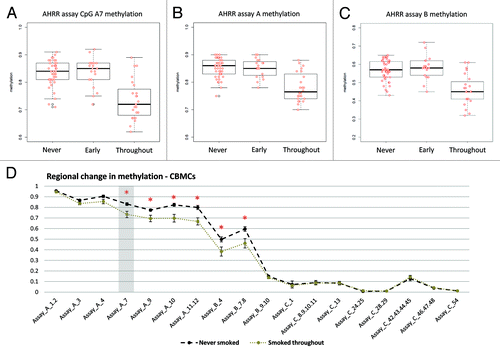
Table 2. DNA methylation level across the AHRR gene body region
Furthermore, we measured methylation at the GFI1 and MYO1G genes, which showed significant differences in response to smoking in the Joubert study,Citation10 but was not identified as significant in adult studies.Citation11-Citation14 Our assay targeted the region reported to be the most differentially methylated in both genes and several surrounding CpG sites.We found no evidence for an effect of maternal methylation in either case (Figs. S4 and S5). This is in agreement with other HM450-based smoking studies in adults,Citation11-Citation14 though it should be noted that two different CpG sites within GFI1 were associated with smoking by Zeilinger et al.Citation14
Association between AHRR intron 3 methylation and AHRR expression
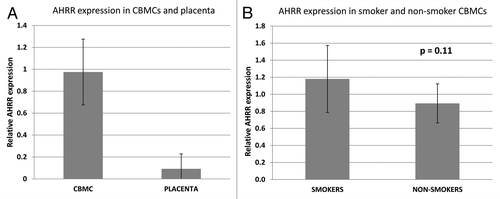
The enrichment of active histone marks and the tissue-specific methylation pattern within the smoking associated differentially methylated region, suggest that it is functionally important. Using expression primers previously published by Shenker et al.,Citation12 we analyzed AHRR expression in CBMCs and placentas from smoking and non-smoking mothers. As expected, AHRR expression was several folds higher in CBMCs compared with placenta (), and there was a non-significant trend toward higher expression in CBMCs exposed to smoke ().
Figure 3. AHRR expression in CBMCs exposed and not exposed to smoke during pregnancy. (A) AHRR expression is higher in CBMCs compared with placenta tissue. (B) AHRR expression in CBMCs exposed to smoking during pregnancy (n = 5) is slightly higher than in non-smoking controls (n = 10), P = 0.11. Bars represent standard deviation.
Maternal smoking-induced hypomethylation of AHRR is maintained at 18 mo
We next analyzed DNA methylation in matched 18-mo peripheral blood mononuclear cells (PBMCs), to determine if the DNA methylation changes within the AHRR gene body persist after birth. Due to the small number of longitudinally collected bloods from twins of mothers that “smoked throughout,” only 3 unrelated individuals with matched birth and 18 mo mononuclear cells were available for analysis in this group, along with 8 “never smoked” control individuals (Fig. S6). shows the mean methylation for birth and 18 mo individuals in “smoked” and “never” groups. Interestingly, both groups showed an increase in mean methylation from birth to 18 mo; however, the difference in mean methylation between the “smoked throughout” and “never smoked” remained significantly different (; ), albeit at a slightly reduced level on average. (; Fig. S7). Interestingly, none from the “smoked throughout” pregnancy group analyzed longitudinally reported “household smoking” after birth, this suggests that the persistent differences in methylation were due to smoking exposure during pregnancy, and were not due to continued second hand exposure to smoking after birth. The absence of plasma cotinine in infants at 18 mo supports this conclusion (Table S2).
Figure 4. Maternal smoking associated DNA methylation change is maintained in peripheral blood mononuclear cells at 18 mo of age. DNA methylation levels (y-axis) across assay A and B at birth and 18 mo in smokers and controls. There is a general increase in methylation, at the majority smoking-associated CpG sites, from birth to 18 mo in both groups. DNA methylation differences between smoked throughout and never smoked groups remain significant at several CpG sites, however the absolute difference in methylation between the groups is lower at 18 mo.
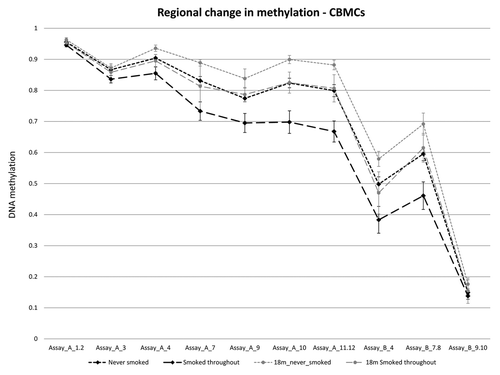
Table 3. DNA methylation at 18 mo in smokers and never smoked controls
Within-pair analysis of MZ and DZ twins suggests a role for genetic factors in regulating DNA methylation at the AHRR gene body
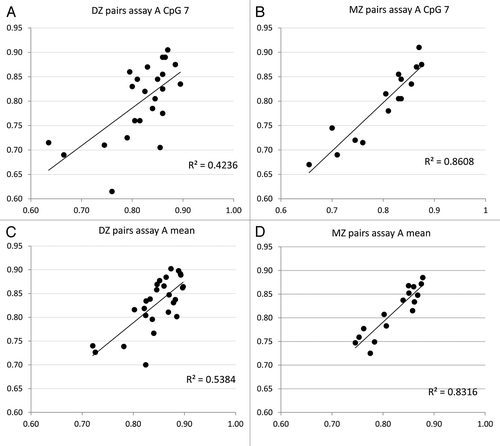
We have previously utilized the twin model to examine the genetic and environmental influence on DNA methylation at specific genes and on a genome-scale level,Citation18-Citation20 and found clear evidence for both genetic and cumulative environmental/stochastic factors in contributing to the neonatal epigenetic profile. By comparing within-pair DNA methylation similarity at the AHRR gene between monozygotic (MZ) and dizygotic (DZ) twins, the contribution of underlying genetic variation to DNA methylation at a particular locus could be estimated. Linear regression analysis of methylation levels at CpG_A7, or the mean of assay A, revealed that MZ twins as a group are generally more similar (R2 = 0.86 and 0.83) in their DNA methylation level than DZ twins (R2 = 0.42 and 0.54) at birth ().
Figure 5. Twin model analysis suggests a genetic component to AHRR methylation. The classic twin model analysis was performed to determine the genetic contribution to AHRR methylation at CpG_A7 and across assay A. Twin “1” methylation level is shown on the x axis, and twin “2” methylation level on the y-axis. The within-pair correlation (R2) was higher in MZ twins (B and D) compared with DZ twins (A and C). This suggests that MZ twins are generally more similar in their AHRR DNA methylation level than DZ twins, indicating a genetic control of methylation at this locus.
Discussion
Several in utero environments have been linked to changes in neonatal epigenetic profile (reviewed extensively in Hogg et al.Citation21). Many of these epigenetic studies were facilitated by the establishment and maintenance of large longitudinal birth cohorts that are now beginning to establish the case for epigenetic marks as the mediators of DOHaD mechanisms.Citation6 It is important to note that an epigenetic response to an in utero exposure need not always be detrimental and may in fact be a protective adaptation with the potential to confer beneficial outcomes to the progeny, or even future generations. The latter has been reported in a rat model of carbon tetrachloride-induced liver damage, associated with epigenetically mediated increased hepatic healing. Of particular note, this effect was amplified transgenerationally, such that exposure in the third generation was not associated with significant liver damage.Citation22 However, replication of many reported findings is generally lacking.
In order to definitively show a role of epigenetics in “programming” later health risk in humans in accordance with the DOHaD hypothesis, several requirements need to be met. First, an association between a specific intrauterine environment and a specific epigenetic change must be established, which can be difficult given the plethora of confounding factors of genetic, environmental, stochastic, and temporal origin.Citation23 Next, these results need to be reproducible across cohorts of similar ethnicity. In most instances, it is likely that an epigenetic change detected at birth needs to be stable over time, and must be reproducibly associated with the later onset of a specific complex disease. Finally, direct evidence for a functional role of the associated epigenetic change needs to be obtained. Given the long latency of many complex disease phenotypes implicated in DOHaD, these prerequisites are unlikely to be firmly established for many human disorders for some time due to a lack of suitable mature longitudinal cohorts with multiple biospecimen collections.
Replication of results has been a major issue for the field, recently highlighted for six studies that looked at the relationship between methylation and birth weight, each of which reported completely non-overlapping sets of genes.Citation24 The disparity may be due to differences in study design (e.g., twin or singleton), differences in birth weight range and discordance between individuals, cells analyzed, or genetic background of each cohort. It is therefore of utmost importance to attempt to replicate results in a timely manner, so as to guide future functional studies, and to eliminate false positives. To date, the association between tobacco smoking exposure and DNA methylation within the AHRR gene remains the most convincing example of the relationship between a specific environment and DNA methylation differences in humans.Citation10-Citation14
AHRR is a negative regulator of the AHR (aryl hydrocarbon receptor) gene, which codes for a protein that binds to a wide range of xenobiotics including nicotine and caffeine.Citation25 AHR induces the expression of CYP1A1 and other genes involved in the removal of deleterious chemicals, while also playing a role in a range of other cellular pathways, including cell cycle control.Citation26 AHRR exerts its repressive action on AHR by binding to ARNT, a partner of AHR, and therefore higher AHRR expression associated with gene hypomethylation might be anticipated to attenuate the cellular responses to smoking. However, it has not been conclusively shown that higher AHRR expression is directly associated with lower CYP1A1 expression (discussed in Harper et al.Citation27). Interestingly, Joubert et al. identified opposite DNA methylation changes at AHRR and CYP1A1 in response to smoking, suggesting that both an increase in AHRR and a decrease in CYP1A1 expression may be involved in the response.Citation10AHRR may also serve as a tumor suppressor gene, with evidence for decreased expression in several human tumor types, with a concomitant increase in promoter methylation.Citation28 The role of gene body methylation in the expression of this gene remains unclear, yet the presence of specific chromatin “signatures” at this region supports a functional role in gene regulation. This is supported by concomitant loss of methylation and higher expression of AHRR observed in blood and lungs from adult smokersCitation11,Citation12 It is unclear if AHR and CYP1A1 expression are lowered in lungs of smokers.
In this study we sought to replicate the DNA methylation differences in blood mononuclear cells, and to determine if other cell types are sensitive to maternal smoking in pregnancy. Our data supports previous findings, with several CpG sites within the analyzed region of AHRR showing lower methylation in CBMCs in association with maternal smoking (). Furthermore, we did not identify a relationship between GFI1 and MYO1G methylation and smoking (Figs. S4 and S5). Based on the magnitude of methylation changes observed in the Joubert et al.Citation10 study, our study is underpowered to detect the small methylation differences observed for MYO1G, which may explain why we failed to find a significant association with this gene. On the other hand, our study had over 90% power to detect an association between smoking and GFI1 methylation levels in CBMCs, based on the previously reported effect size.Citation10 This further highlights the need for replication of studies in different cohorts to eliminate false positive and population-specific results.
Analysis of buccal epithelium and placenta did not identify a smoking-associated methylation difference at AHRR in these tissues (Fig. S2). Interestingly, this region of differential methylation also showed a tissue-specific pattern, with CBMCs showing high methylation across the region (approximately 80%), while buccal epithelium and placenta showed significantly lower methylation across several CpG sites (40–70%; ). This further suggests that the intron 3 of AHRR harbors a regulatory region, but this will require direct testing. Previous data suggest that AHRR is expressed at low levels in the placenta, while cord blood mononuclear cells show high interindividual variation, with no/low expression to very high expression.Citation29 Unfortunately, previous analysis did not explore the relationship between maternal smoking during pregnancy and AHRR expression in CBMCs. However, higher AHRR expression has previously been detected in lungs of smokers and lung cancer patients with a history of smoking.Citation12,Citation30 Our expression analysis identified a trend toward higher expression in CBMCs exposed to smoking during pregnancy (P = 0.11; ). A weakness of this analysis was the low number of individuals for which CBMC RNA was available (n = 5). Of these, only one sample showed a large drop in methylation at the AHRR intron 3 region. Therefore, our results suggest that smoking may not alter AHRR expression in CBMCs to the extent it does in lungs of patients, although this will require further testing in a larger sample set. It would be much more informative, however, to examine expression in CBMCs from individuals with a larger loss of methylation at intron 3 or to directly test for this association using appropriate in vitro reporter assays.
Another aspect of the relationship between the maternal environment and fetal epigenetics we wanted to explore was the critical time for an environment to affect DNA methylation during pregnancy. In our cohort, we analyzed a subset of women that smoked pre/peri-conception (i.e., “smoked early”), but not after. There was no difference in DNA methylation between the “never smoked” and the “smoked early” groups (). This suggests that early fetal exposure to maternal smoking is not sufficient to induce a DNA methylation change that is measurable at birth, at least in the tissues we examined. Furthermore, none of the mothers in the cohort smoked in the third trimester, without also smoking earlier in pregnancy, so it is difficult to pinpoint the critical period for when smoking induces DNA methylation differences. Our data support a mechanism whereby prolonged exposure is necessary to induce a detectable long lasting change in methylation. Alternatively, it is possible that short-medium term exposure later in pregnancy, possibly at a particularly sensitive point in fetal blood development, can induce a change in methylation at the AHRR locus. Furthermore, we did not identify a sex effect on DNA methylation level or response to maternal smoking (), which has previously been reported for some maternal environments in humans and mice.Citation31,Citation32
In accordance with previous reports,Citation10 we found a strong correlation between self-reported questionnaire data on smoking behavior and plasma cotinine levels, both before and after pregnancy. Furthermore, our questionnaire data and plasma cotinine levels suggest that none of the twins was exposed to household smoking after birth, suggesting that the lower methylation observed at 18 mo is an epigenetic “legacy” of intrauterine exposure. Our data are in accordance with a recent adult study that showed that AHRR DNA methylation in adult blood of smokers that quit approaches the levels of never smokers within the first few years of quitting, but never completely reaches normal levels, remaining on average 3–4% lower.Citation14 Despite the small numbers of samples available, we found evidence of a consistent effect that showed statistical significance (; ). It will also be interesting to study the effects of paternal only postnatal smoking exposure, on AHRR methylation in children.
In conclusion, we have confirmed the association of maternal smoking throughout pregnancy and decreasing AHRR methylation in blood at birth. We have expanded the region of interest, and show that the smoking-associated DNA methylation changes are limited to the CpG island shore, and are tissue-specific. Finally, we show evidence that intrauterine exposure can induce persistent DNA methylation change up to 18 mo of age. Given the role of AHRR as a negative regulator of pathways involved in pleiotropic responses to environmental contaminants, the observed “programming” of this gene in utero following exposure to maternal smoking is likely to be of functional relevance. At present, however, it is unclear whether the observed response to smoking is an adaptive change associated with postnatal benefit, whether it is neutral, or potentially detrimental to postnatal health. This is the first step toward providing a link between environmental exposures in utero, epigenetic disruption, and DOHaD. Further analysis in a larger longitudinal birth cohort is necessary to confirm these results.
Methods
Samples
The Peri/postnatal Epigenetics Twin Study (PETS) is a cohort of 251 young twins and their mothers, recruited mid-way through pregnancy, for which lifestyle/environment data and tissue samples were collected at birth and at 18 mo of age.Citation15,Citation16 The mothers were predominantly of white European descent, and women with poor English language skills were not enrolled into the study. For the current study, we used cord blood mononuclear cells (CBMCs), buccal (inner cheek) epithelium, and placenta tissue collected at birth. Maternal smoking data from questionnaires (including number of cigarettes per week) covered pre/peri-conception (prior to knowledge of the pregnancy), from known to 12 wk, 12–24 wk, and 24–36 wk of gestation, and the presence or absence of household smoking in the first 18 mo of life. Cases were mothers who reported any smoking during these time points and controls were mothers who reported never smoking and were matched to the smoking group for gestational age, maternal BMI, birth weight, birth weight discordance, and serum folate concentration at 28 wk gestation (). Of a total 251 pregnant women recruited in our original study, 12 reported smoking throughout pregnancy, while 11 reported smoking only in early pregnancy up to finding out they were pregnant. CBMCs were collected from all of these pregnancies (46 babies). Additionally, CBMCs from 23 matched pregnancies, with no reported smoking were also analyzed as controls (). In addition, we analyzed CBMCs from 23 matched pregnancies, with no reported smoking before or during pregnancy (). Cotinine levels were measured in maternal plasma at 28 wk and infant plasma at birth and 18 mo for 6 twin pairs for which samples and methylation data were available at birth and 18 mo. Cotinine measurements were performed with HPLC (Agilent), using a protocol adapted from Kellogg et al.Citation33
DNA methylation quantification
DNA extraction from cells and tissue was performed using the phenol/chloroform method, as previously described.Citation20 Genomic DNA was bisulfite treated using the MethylEasy DNA conversion kit (Human Genetic Signatures). DNA methylation was quantified using the SEQUENOM MassARRAY EpiTYPER platform, as previously described.Citation20,Citation34,Citation35 Duplicate PCRs were performed, and both PCR products were analyzed using the EpiTYPER platform. To minimize technical variation, replicates that showed methylation difference greater than 0.1 (10%) were removed from analysis. Assays were designed using the EpiDesigner software (www.epidesigner.com) and cleavage patterns were determined using the “ampliconPrediction” function in R. All forward primers contain a balance tag (AGGAAGAGAG) and all reverse primers contain a T7 tag (CAGTAATACG ACTCACTATA GGGAGAAGGC T). Primers used were: Assay A F- 5′ GTTGGTAATG GTTTTGAGAT TTT 3′ R- 5′ AAAACCAACC TATCCCCTAC CTC 3′ Assay B: F- 5′ AGTGGTTTTG GTAGGGTTTT TTTT 3′ R- 5′ AACCCCAATC TCCTCCTCTA TAAT 3′ and Assay C: F- 5′ TTTAGGTGGG ATTTTTAGGT TTAGG 3′ R- 5′ ATACAACCAA ACCCCATTAC AAAA 3′. Assay information including primer sequences is shown in and all methylation values for CBMCs are shown in Table S1.
Gene expression analysis
RNA from CBMCs and placenta was reverse transcribed using the Tetro cDNA synthesis kit (Bioline). AHRR expression was quantified using a previously published assay,Citation12 on the Roche lightcycler with the Sensifast qPCR mix (Bioline). Relative expression was calculated using the delta delta Ct method.
Data analysis
The freely available R statistical program (cran.r-project.org/) was used to produce descriptive figures, including heatmaps and box-and-whisker plots, and to determine the suitability of EpiTYPER assays to cover CpG sites of interest. Differences in methylation level between the different groups were determined using the Student t test. The classical twin model approach was used to determine the genetic contribution to DNA methylation level. The model is based on the fact that monozygotic (MZ) twins are genetically identical, while dizygotic (DZ) twins share about 50% of their genetic variation. DNA methylation was treated as a continuous variable, and the within-pair correlation was calculated using linear regression. Higher similarity in DNA methylation profile in MZ twins as a group relative to DZ twins as a group is supportive of a role of genetic influence on methylation levels.
| Abbreviations: | ||
| AHRR | = | aryl hydrocarbon receptor repressor |
| CBMC | = | cord blood mononuclear cells |
| DOHaD | = | developmental origins of health and disease |
| HM450 | = | Infinium HumanMethylation450 BeadChip |
Additional material
Download Zip (1.5 MB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We wish to thank Ruth Morley, John Carlin, Mark Umstad, Euan Wallace, and Michael Permezel for their contributions to establishing the PETS cohort; Sarah Healy, Tina Vaiano, Nicole Brooks, Jennifer Foord, Sheila Holland, Anne Krastev, Siva Illancheran, and Joanne Mockler for recruitment and sample collection; Xin Li, Ji Hoon E Joo, Anna Czajko, Geraldine McIlroy, and all mothers and twins that participated in this study. We also thank Ben Ong for help with Sequenom analysis and Megan Thompson for technical work. This work was supported by grants from the from the Australian National Health and Medical Research Council (grant numbers 437015 and 607358 to Craig JM and Saffery R), the Financial Markets Foundation for Children (grant no. 032-2007), and by the Victorian Government's Operational Infrastructure Support Program. Saffery R is supported by a NHMRC Senior Research Fellowship. Craig JM would also like to acknowledge financial support from the Murdoch Childrens Research Institute. Ryan J is supported by an NHMRC Training (Postdoctoral) Fellowship (Overseas Public Health, APP1012735).
References
- Barker DJ. The fetal and infant origins of adult disease. BMJ 1990; 301:1111; http://dx.doi.org/10.1136/bmj.301.6761.1111; PMID: 2252919
- Barker DJ. Intra-uterine programming of the adult cardiovascular system. Curr Opin Nephrol Hypertens 1997; 6:106 - 10; http://dx.doi.org/10.1097/00041552-199701000-00017; PMID: 9051361
- Barker DJ. The developmental origins of insulin resistance. Horm Res 2005; 64:Suppl 3 2 - 7; http://dx.doi.org/10.1159/000089311; PMID: 16439838
- Barker DJ, Winter PD, Osmond C, Margetts B, Simmonds SJ. Weight in infancy and death from ischaemic heart disease. Lancet 1989; 2:577 - 80; http://dx.doi.org/10.1016/S0140-6736(89)90710-1; PMID: 2570282
- Roseboom TJ, van der Meulen JH, Ravelli AC, Osmond C, Barker DJ, Bleker OP. Effects of prenatal exposure to the Dutch famine on adult disease in later life: an overview. Mol Cell Endocrinol 2001; 185:93 - 8; http://dx.doi.org/10.1016/S0303-7207(01)00721-3; PMID: 11738798
- Waterland RA, Michels KB. Epigenetic epidemiology of the developmental origins hypothesis. Annu Rev Nutr 2007; 27:363 - 88; http://dx.doi.org/10.1146/annurev.nutr.27.061406.093705; PMID: 17465856
- Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer 2004; 4:143 - 53; http://dx.doi.org/10.1038/nrc1279; PMID: 14732866
- Ptak C, Petronis A. Epigenetics and complex disease: from etiology to new therapeutics. Annu Rev Pharmacol Toxicol 2008; 48:257 - 76; http://dx.doi.org/10.1146/annurev.pharmtox.48.113006.094731; PMID: 17883328
- Rakyan VK, Down TA, Balding DJ, Beck S. Epigenome-wide association studies for common human diseases. Nat Rev Genet 2011; 12:529 - 41; http://dx.doi.org/10.1038/nrg3000; PMID: 21747404
- Joubert BR, Håberg SE, Nilsen RM, Wang X, Vollset SE, Murphy SK, Huang Z, Hoyo C, Midttun Ø, Cupul-Uicab LA, et al. 450K epigenome-wide scan identifies differential DNA methylation in newborns related to maternal smoking during pregnancy. Environ Health Perspect 2012; 120:1425 - 31; http://dx.doi.org/10.1289/ehp.1205412; PMID: 22851337
- Monick MM, Beach SR, Plume J, Sears R, Gerrard M, Brody GH, Philibert RA. Coordinated changes in AHRR methylation in lymphoblasts and pulmonary macrophages from smokers. Am J Med Genet B Neuropsychiatr Genet 2012; 159B:141 - 51; http://dx.doi.org/10.1002/ajmg.b.32021; PMID: 22232023
- Shenker NS, Polidoro S, van Veldhoven K, Sacerdote C, Ricceri F, Birrell MA, Belvisi MG, Brown R, Vineis P, Flanagan JM. Epigenome-wide association study in the European Prospective Investigation into Cancer and Nutrition (EPIC-Turin) identifies novel genetic loci associated with smoking. Hum Mol Genet 2013; 22:843 - 51; http://dx.doi.org/10.1093/hmg/dds488; PMID: 23175441
- Philibert RA, Beach SR, Brody GH. Demethylation of the aryl hydrocarbon receptor repressor as a biomarker for nascent smokers. Epigenetics 2012; 7:1331 - 8; http://dx.doi.org/10.4161/epi.22520; PMID: 23070629
- Zeilinger S, Kühnel B, Klopp N, Baurecht H, Kleinschmidt A, Gieger C, Weidinger S, Lattka E, Adamski J, Peters A, et al. Tobacco smoking leads to extensive genome-wide changes in DNA methylation. PLoS One 2013; 8:e63812; http://dx.doi.org/10.1371/journal.pone.0063812; PMID: 23691101
- Loke YJ, Novakovic B, Ollikainen M, Wallace EM, Umstad MP, Permezel M, Morley R, Ponsonby AL, Gordon L, Galati JC, et al. The Peri/postnatal Epigenetic Twins Study (PETS). Twin Res Hum Genet 2013; 16:13 - 20; http://dx.doi.org/10.1017/thg.2012.114; PMID: 23171547
- Saffery R, Morley R, Carlin JB, Joo JH, Ollikainen M, Novakovic B, Andronikos R, Li X, Loke YJ, Carson N, et al. Cohort profile: The peri/post-natal epigenetic twins study. Int J Epidemiol 2012; 41:55 - 61; http://dx.doi.org/10.1093/ije/dyr140; PMID: 22422448
- Das R, Lee YK, Strogantsev R, Jin S, Lim YC, Ng PY, Lin XM, Chng K, Yeo GSh, Ferguson-Smith AC, et al. DNMT1 and AIM1 Imprinting in human placenta revealed through a genome-wide screen for allele-specific DNA methylation. BMC Genomics 2013; 14:685; http://dx.doi.org/10.1186/1471-2164-14-685; PMID: 24094292
- Gordon L, Joo JE, Powell JE, Ollikainen M, Novakovic B, Li X, Andronikos R, Cruickshank MN, Conneely KN, Smith AK, et al. Neonatal DNA methylation profile in human twins is specified by a complex interplay between intrauterine environmental and genetic factors, subject to tissue-specific influence. Genome Res 2012; 22:1395 - 406; http://dx.doi.org/10.1101/gr.136598.111; PMID: 22800725
- Novakovic B, Galati JC, Chen A, Morley R, Craig JM, Saffery R. Maternal vitamin D predominates over genetic factors in determining neonatal circulating vitamin D concentrations. Am J Clin Nutr 2012; 96:188 - 95; http://dx.doi.org/10.3945/ajcn.112.035683; PMID: 22648713
- Ollikainen M, Smith KR, Joo EJ, Ng HK, Andronikos R, Novakovic B, Abdul Aziz NK, Carlin JB, Morley R, Saffery R, et al. DNA methylation analysis of multiple tissues from newborn twins reveals both genetic and intrauterine components to variation in the human neonatal epigenome. Hum Mol Genet 2010; 19:4176 - 88; http://dx.doi.org/10.1093/hmg/ddq336; PMID: 20699328
- Hogg K, Price EM, Hanna CW, Robinson WP. Prenatal and perinatal environmental influences on the human fetal and placental epigenome. Clin Pharmacol Ther 2012; 92:716 - 26; http://dx.doi.org/10.1038/clpt.2012.141; PMID: 23047650
- Zeybel M, Hardy T, Wong YK, Mathers JC, Fox CR, Gackowska A, Oakley F, Burt AD, Wilson CL, Anstee QM, et al. Multigenerational epigenetic adaptation of the hepatic wound-healing response. Nat Med 2012; 18:1369 - 77; http://dx.doi.org/10.1038/nm.2893; PMID: 22941276
- Foley DL, Craig JM, Morley R, Olsson CA, Dwyer T, Smith K, Saffery R. Prospects for epigenetic epidemiology. Am J Epidemiol 2009; 169:389 - 400; http://dx.doi.org/10.1093/aje/kwn380; PMID: 19139055
- Souren NY, Lutsik P, Gasparoni G, Tierling S, Gries J, Riemenschneider M, Fryns JP, Derom C, Zeegers MP, Walter J. Adult monozygotic twins discordant for intra-uterine growth have indistinguishable genome-wide DNA methylation profiles. Genome Biol 2013; 14:R44; http://dx.doi.org/10.1186/gb-2013-14-5-r44; PMID: 23706164
- Denison MS, Nagy SR. Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Annu Rev Pharmacol Toxicol 2003; 43:309 - 34; http://dx.doi.org/10.1146/annurev.pharmtox.43.100901.135828; PMID: 12540743
- Elizondo G, Fernandez-Salguero P, Sheikh MS, Kim GY, Fornace AJ, Lee KS, Gonzalez FJ. Altered cell cycle control at the G(2)/M phases in aryl hydrocarbon receptor-null embryo fibroblast. Mol Pharmacol 2000; 57:1056 - 63; PMID: 10779392
- Harper PA, Riddick DS, Okey AB. Regulating the regulator: factors that control levels and activity of the aryl hydrocarbon receptor. Biochem Pharmacol 2006; 72:267 - 79; http://dx.doi.org/10.1016/j.bcp.2006.01.007; PMID: 16488401
- Zudaire E, Cuesta N, Murty V, Woodson K, Adams L, Gonzalez N, Martínez A, Narayan G, Kirsch I, Franklin W, et al. The aryl hydrocarbon receptor repressor is a putative tumor suppressor gene in multiple human cancers. J Clin Invest 2008; 118:640 - 50; PMID: 18172554
- Yamamoto J, Ihara K, Nakayama H, Hikino S, Satoh K, Kubo N, Iida T, Fujii Y, Hara T. Characteristic expression of aryl hydrocarbon receptor repressor gene in human tissues: organ-specific distribution and variable induction patterns in mononuclear cells. Life Sci 2004; 74:1039 - 49; http://dx.doi.org/10.1016/j.lfs.2003.07.022; PMID: 14672759
- Bossé Y, Postma DS, Sin DD, Lamontagne M, Couture C, Gaudreault N, Joubert P, Wong V, Elliott M, van den Berge M, et al. Molecular signature of smoking in human lung tissues. Cancer Res 2012; 72:3753 - 63; http://dx.doi.org/10.1158/0008-5472.CAN-12-1160; PMID: 22659451
- Jensen Peña C, Monk C, Champagne FA. Epigenetic effects of prenatal stress on 11β-hydroxysteroid dehydrogenase-2 in the placenta and fetal brain. PLoS One 2012; 7:e39791; http://dx.doi.org/10.1371/journal.pone.0039791; PMID: 22761903
- Gabory A, Ferry L, Fajardy I, Jouneau L, Gothié JD, Vigé A, Fleur C, Mayeur S, Gallou-Kabani C, Gross MS, et al. Maternal diets trigger sex-specific divergent trajectories of gene expression and epigenetic systems in mouse placenta. PLoS One 2012; 7:e47986; http://dx.doi.org/10.1371/journal.pone.0047986; PMID: 23144842
- Kellogg MD, Behaderovic J, Bhalala O, Rifai N. Rapid and simple tandem mass spectrometry method for determination of serum cotinine concentration. Clin Chem 2004; 50:2157 - 9; http://dx.doi.org/10.1373/clinchem.2004.039594; PMID: 15502087
- Ehrich M, Nelson MR, Stanssens P, Zabeau M, Liloglou T, Xinarianos G, Cantor CR, Field JK, van den Boom D. Quantitative high-throughput analysis of DNA methylation patterns by base-specific cleavage and mass spectrometry. Proc Natl Acad Sci U S A 2005; 102:15785 - 90; http://dx.doi.org/10.1073/pnas.0507816102; PMID: 16243968
- Wong NC, Novakovic B, Weinrich B, Dewi C, Andronikos R, Sibson M, Macrae F, Morley R, Pertile MD, Craig JM, et al. Methylation of the adenomatous polyposis coli (APC) gene in human placenta and hypermethylation in choriocarcinoma cells. Cancer Lett 2008; 268:56 - 62; http://dx.doi.org/10.1016/j.canlet.2008.03.033; PMID: 18485586