
Abstract
DNA methyltransferase 1 (DNMT1) is essential for DNA methylation, gene regulation and chromatin stability. We previously discovered DNMT1 mutations cause hereditary sensory and autonomic neuropathy type 1 with dementia and hearing loss (HSAN1E; OMIM 614116). HSAN1E is the first adult-onset neurodegenerative disorder caused by a defect in a methyltransferase gene. HSAN1E patients appear clinically normal until young adulthood, then begin developing the characteristic symptoms involving central and peripheral nervous systems. Some HSAN1E patients also develop narcolepsy and it has recently been suggested that HSAN1E is allelic to autosomal dominant cerebellar ataxia, deafness, with narcolepsy (ADCA-DN; OMIM 604121), which is also caused by mutations in DNMT1. A hotspot mutation Y495C within the targeting sequence domain of DNMT1 has been identified among HSAN1E patients. The mutant DNMT1 protein shows premature degradation and reduced DNA methyltransferase activity. Herein, we investigate genome-wide DNA methylation at single-base resolution through whole-genome bisulfite sequencing of germline DNA in 3 pairs of HSAN1E patients and their gender- and age-matched siblings. Over 1 billion 75-bp single-end reads were generated for each sample. In the 3 affected siblings, overall methylation loss was consistently found in all chromosomes with X and 18 being most affected. Paired sample analysis identified 564,218 differentially methylated CpG sites (DMCs; P < 0.05), of which 300 134 were intergenic and 264 084 genic CpGs. Hypomethylation was predominant in both genic and intergenic regions, including promoters, exons, most CpG islands, L1, L2, Alu, and satellite repeats and simple repeat sequences. In some CpG islands, hypermethylated CpGs outnumbered hypomethylated CpGs. In 201 imprinted genes, there were more DMCs than in non-imprinted genes and most were hypomethylated. Differentially methylated region (DMR) analysis identified 5649 hypomethylated and 1872 hypermethylated regions. Importantly, pathway analysis revealed 1693 genes associated with the identified DMRs were highly associated in diverse neurological disorders and NAD+/NADH metabolism pathways is implicated in the pathogenesis. Our results provide novel insights into the epigenetic mechanism of neurodegeneration arising from a hotspot DNMT1 mutation and reveal pathways potentially important in a broad category of neurological and psychological disorders.
Introduction
DNA methylation affects many cellular functions including transcription regulation, cell differentiation, gene imprinting and genome stability.Citation1,Citation2 Mammalian DNA methylation is catalyzed by DNMT1, DNMT3A, and DNMT3B. DNA methyltransferase1 (DNMT1), locates at chromosome 19p13.3-p13.2, is the maintenance methyltransferase and essential for maintaining proper epigenetic inheritance. DNMT3A and DNMT3B establish new methylation patterns and are considered de novo methyltransferases. The line between maintenance and de novo methyltransferases has become increasingly blurred as studies have linked DNMT1 to de novo methylationCitation3 and DNMT3A/B to maintenance methylation.Citation4 Aberrant DNA methylation and other epigenetic defects by either Mendelian or polygenic pathogenesis are increasingly associated with diverse forms of neurological diseases.Citation1,Citation5,Citation6 DNMT3B mutation leading to the partial inactivation of its catalytic domain has been linked to a human developmental disorder, ICF1 (immunodeficiency, centromere instability, and facial anomalies) syndrome.Citation7 DNMT3B mutations cause DNA hypomethylation in satellites 2 and 3 and α repetitive sequences, a major part of constitutive heterochromatin. Recently, we identified heterozygous mutations in DNMT1 targeting sequence (TS) domain causing hereditary sensory and autonomic neuropathy type 1 with dementia and hearing loss (HSAN1E; OMIM 614116).Citation8 Subsequently, additional DNMT1 TS domain mutations were found underlying autosomal dominant cerebellar ataxia, deafness and narcolepsy (ADCA-DN, OMIM 604121).Citation9 It has been suggested that these two adult-onset neurodegenerative disorders can be considered in the differential of each other as HSAN1E and ADCA-DN patients may share a similar set of phenotypes with varied severity and onset age.Citation10 More HSAN1E families have been recognized and a hot spot mutation at amino acid 495 was identified as Y495C mutation is the most common causal mutation found among HSAN1E patients.Citation8,Citation11 The DNMT1 Y495C mutation leads to DNMT1 protein misfolding and decreased enzymatic activity. DNMT1 is indispensable for development, cellular functions and cell cycle regulation. Genomic deletion of DNMT1 in mice leads to embryonic death,Citation12 and complete deletion of DNMT1 in human cancer cells results in significant loss of global methylation, pronounced chromosomal defects and apoptosis.Citation13 Dynamic changes in DNA methylation are also linked to neuronal synaptic stimulation in the brain.Citation14 DNMT1 mutations in HSAN1E do not lead to malignancy; instead, they result in the neurodegeneration of peripheral and central nervous systems.Citation8,Citation15
The aim of the present study was to investigate the genome-wide DNA methylation changes in HSAN1E patients arising from the hotspot DNMT1 mutation Y495C. There are ~30 million cytosines preceding guanine nucleotides (CpGs)Citation16 in the human genome and the majority of them (~80%) are methylated,Citation2 especially at repetitive elements and centromeric satellite repeats, which comprise approximately half of human genome. A small percentage of CpG dinucleotides are clustered within gene promoters as CpG islands (CGI), but normally only 3% of CpG islands are methylated.Citation17 The pathogenic role of promoter methylation has been extensively studied especially in cancer, but there is limited understanding on how methylation changes in intergenic regions relate to human disease.
We analyzed CpG methylation at single-base resolution in 3 affected individuals and their age-, gender-matched (<5 y difference) unaffected siblings by whole genome bisulfite sequencing (WGBS). This paired-sample study design allowed us to perform statistical analysis of differential methylation while controlling for extraneous factors, minimizing inter-familial and environmental influences. Furthermore, we investigated the pathways downstream of consequent aberrant DNA methylation that are involved in HSAN1E pathogenesis in relation to other neurological and psychological disorders.
Results
Genomic DNA was extracted from peripheral blood B cells of three affected patients carrying the heterozygous Y495C DNMT1 mutation and their unaffected siblings and analyzed by WGBS and bioinformatics approaches as illustrated in . Our sequencing results generated approximately one billion single-end reads of 75-bp length for each individual sample. Alignment efficiency was high with 94–95% of these reads mapped to the human reference genome (hg19; Table 1). Our sequencing results covered ~24 of the ~30 million CpGs present in the human genome. The bisulfite conversion rate was assessed using all non-CpG sites with > 10X coverage in the genome, and found to be close to 1 (>0.999), indicating almost complete conversion. On average, 18–19 million of CpG cytosines were obtained with >10X coverage for each individual genome and 12 035 253 CpG sites had >10X coverage across all 6 samples (), of which 6 051 917 were in genic (5kb upstream or inside gene body) regions and 5 983 334 were in intergenic (outside of genic regions) regions. The genic CpGs had higher overall methylation () than the intergenic CpGs (). Notably, the affected patients consistently had lower methylation than their paired siblings, particularly in intergenic regions. The methylation of CpG islands (CGI) was low (~30%) across all samples, whereas methylation in repeat regions (Alu, L1, L2, Satellite repeat, and simple repeat) was high (≥70%) (). All the repeat regions had lower methylation in the affected patients than their unaffected siblings except for CGIs ().
Figure 1. Study design and analysis workflow. Three pairs of affected (with DNMT1 mutation and HSAN1) and unaffected siblings (without DNMT1 mutation and HSAN1) were selected from three different families of the same kinship. The sibling pairs were matched for age (<5 y) and gender. WGBS was performed on peripheral lymphocyte cells and DMCs and DMRs were identified by paired analyses. Genes with DMRs 5 kb from theTSS were used for pathway enrichment analysis. A1-A3, affected individuals with DNMT1 mutation; U1-U3, unaffected siblings without DNMT1 mutation.
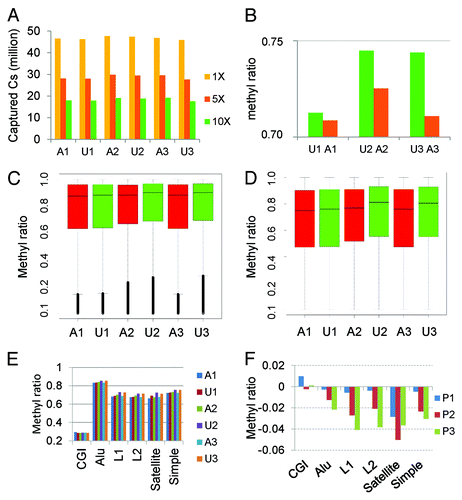
Figure 2. Genome coverage and overall methylation patterns. (A) Captured Cs in CpG context at different depths of coverage. Almost all CpGs in the genome were covered; however, CpGs with at least 10X coverage accounted for about 1/3 of ~30million CpGs in the genome. (B) Overall mean methylation was reduced by 0.3–3% in affected individuals (A1–3) relative to unaffected controls (U1–3;). (C) Methylation distribution of genic CpGs by sample. The affected had slightly lower methylation than their unaffected siblings. The horizontal bar within each box is the median methylation for the sample. (D) Methylation distribution of intergenic CpGs by sample. The affected patients had lower methylation than unaffected individuals and the differences were greater than in genic regions. The horizontal bar within each box is the median methylation for the sample. (E) CGI and repeat region methylation. CGI methylation was low (~30%) across all samples with little difference between samples while methylation in repeat regions (Alu, L1, L2, satellite, and simple repeats) was higher (≥70%). The affected patients consistently had lower methylation (1–5% reduction) than their unaffected siblings. (F) Methylation difference between sibling pairs in CGIs and repeat elements. All repeat elements but not CGIs had lower methylation in the affected individuals with Pair 1 (P1) displaying the smallest differences.
Genome-wide CpG methylation patterns reflect DNMT1 mutation status and kinship
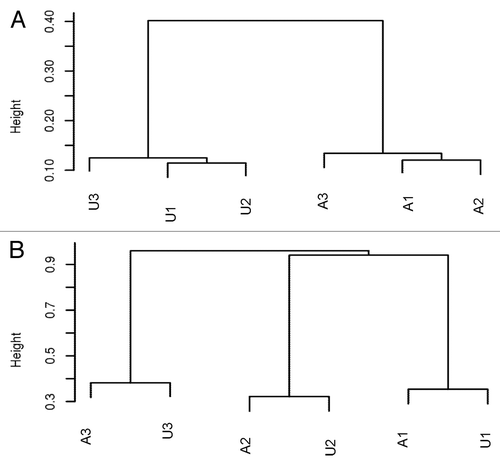
Previous studies have shown that methylation is associated with gender, age, and individual genetic background, as well as environmental influences. Therefore, we designed our study to compare age- and gender-matched sibling pairs to minimize the impact of potentially confounding variables. To validate this approach, we performed unsupervised clustering using the top 126 000 CpGs in which methylation was most varied across the 6 samples (CpGs with the highest standard deviations). We found that the affected and the unaffected samples formed two distinct clusters (), suggesting a strong effect of DNMT1 mutation on DNA methylation. Interestingly, when we used the top 10 000 most varied CpGs across the 6 samples for unsupervised clustering, the samples clustered by relationship (), demonstrating an effect of the overall genetic background. Our results from cluster analysis confirm that it is important to compare the methylation change between gender and age-matched individuals within a family, ideally under similar environmental influence. Also, it indicates that our paired-samples design will likely increase the accuracy in interrogating the impact of DNMT1 mutation on methylation changes of different genomic regions.
Figure 3. (A) Unsupervised clustering using the top 126 000 varied CpGs shows the affected and unaffected samples form different clusters, suggesting the similar methylation patterns by mutation status. (B) Unsupervised clustering using top 10 000 highly varied CpGs shows samples are clustered by family, indicating the strong genetic influence on DNA methylation.
Differentially methylated CpGs (DMCs) are mostly hypomethylated
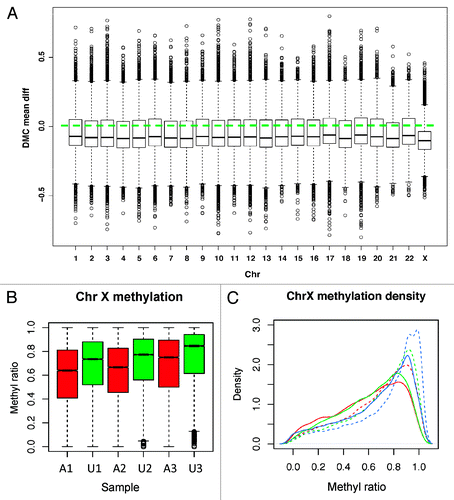
We performed paired t-statistics to identify differentially methylated CpGs or DMCs between 3 affected and 3 unaffected siblings after removing 190 349 CpGs without any variance across samples (constant methylation, non-informative). The mean methylation difference was used to measure the scale of a change along with the differential P value. At P < 0.05, 564 218 DMCs were identified, of which 308 962 had mean ratio difference greater than 0.1 (>10% methylation difference). Noted is that even with paired analysis for increased power, the small sample size would only detect the CpGs with a large difference and low variability. Therefore, we compared the P value distribution from the permutation test with randomly assigned sibling pairs and found that the P values from the true pairs were non-uniform with lower P values shifted to the left side of significant difference (Fig. S1A), whereas the P values from the permutation deviated toward the non-significant side (Fig. S1B). These results indicate that the significant CpGs were not likely random. The majority of the DMCs were hypomethylated in all chromosomes (). Interestingly, chromosome X (3145 hyper- and 10104 hypo-methylated CpGs) and chromosome 18 (4342 hyper- and 11346 hypo-methylated CpGs) had the largest methylation reduction (), suggesting the impact of DNMT1 mutation is chromosome- and region-specific.
Figure 4. Methylation by chromosome. (A) DMCs are plotted for each chromosome separately. All show the dominant hypo-DMCs with chromosome X changed the most. Y-axis: The distribution of methylation mean difference between affected and unaffected individuals. (B) Chromosome X CpG methylation distribution. Affected patients (red) have more obvious reduced methylation than the unaffected (green) for all pairs. (C) Density plot of CpG methylation for chromosome X for each sample. Affected patients have left-shifted (decreased) methylation ratio curves. Solid line: patients with mutation; Dashed lines: sibling controls without mutation. Red lines: A1, A2; Green lines: A2, U2; Blue lines: A3, U3.
Of the 308 962 DMCs with mean methylation change greater than 10%, there were significantly more in intergenic than in genic regions (182 766 vs. 126 196; chi Square test P < 2.2e-16). In genic regions, there were 2-fold more hypomethylated CpGs than hypermethylated CpGs; in intergenic regions, there were 3-fold more hypomethylated CpGs than hypermethylated CpGs (). These results demonstrate higher prevalence of hypomethylation in the intergenic regions. Although the overall, genome-wide reduction in DNA methylation was moderate between affected and unaffected siblings, a greater reduction (5–10%) was observed around transcription start sites (TSS) ().
Figure 5. DMC distribution in different genomic regions. (A) DMCs occur across genomic regions; however, intergenic regions have higher percentage of hypomethylated CpGs than genic regions. (B) Hypomethylation around transcription start site (TSS). All genes are aligned around TSS and average methylation is displayed separately for affected (red) and unaffected (green) subjects. (C) DMCs in different genomic features of genic regions. All regions except CpG islands have more hypo-methylated DMCs in the affected individuals. (D) DMCs in different genomic features of intergenic regions. All have more hypo-methylated DMCs in the affected individuals. X-axis, genomic region; y-axis, percentage of hypermethylated (purple bars) and hypomethylated DMCs (green bars).
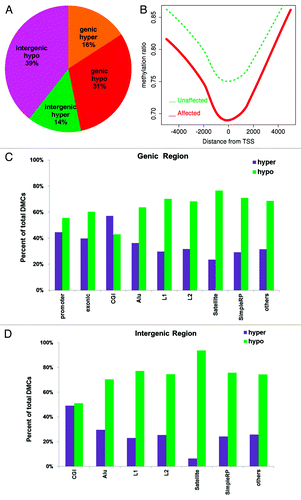
When we assessed the DMC distribution in the genome according to the genomic features such us exons, CGIs, Alu elements, L1, L2, satellite, and simple repeats, we found all but the CGIs to be significantly hypomethylated in both genic () and intergenic regions ().
Differentially methylated regions (DMRs) are mostly intergenic and hypomethylated
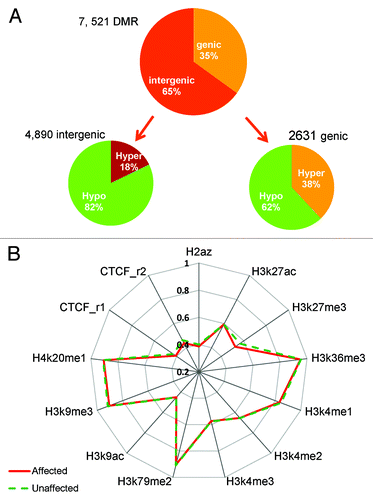
DNA methylation often occurs in clusters and it is important to evaluate how the differentially methylated regions are scattered across the genome. For DMR analysis, we included CpGs captured in all 6 samples and used a smoothing approachCitation18 to stabilize the CpG sites with lower than 10X coverage. We designated a DMR as the minimum of 5 CpGs with the mean difference ≥10% between the 3 pairs of affected and unaffected siblings. Across the genome, 7521 significantly changed DMRs were found, of which 5649 were hypomethylated and 1872 were hypermethylated in the affected siblings. The majority of these DMRs (4890, 65%) were intergenic and 2631 (35%) were within genic regions. Additionally, over 82% of the 4890 intergenic DMRs and 62% of the 2631 genic DMRs were hypomethylated, indicating most DMRs showed loss of methylation in the affected siblings (). The DMR analysis further confirmed more prevalent hypomethylation in the intergenic regions in the affected siblings.
Figure 6. DMR distribution in the genome and DNA methylation in histone marks. (A) Most DMRs are hypomethylated for both genic and intergenic regions; however, this is much more dramatic in intergenic regions (82% vs. 62%). (B) Median methylation difference in different histone mark region between the affected and unaffected individuals. Hypomethylation is observed in H3K27me3, H3K9me3, and CTCF sites.
DMR-associated genes are enriched in neurological disorders
To investigate whether the methylation changes resulted from DNMT1 mutation are directly associated with genes enriched in a particular pathway, functional class or disease, we conducted a pathway enrichment analysis for the genes that have identified DMRs within 5kb of their TSS using Ingenuity Pathway Analysis (IPA; see Methods for details). IPA constructs the networks that optimize for both interconnectivity and number of focus genes under the constraint of maximal network size.Citation19 The top three significantly enriched gene interaction networks were “hereditary disorder, metabolic disease, cancer,” “hereditary disorder, neurological disease, cellular assembly, and organization,” and “cancer, cellular development, tumor morphology.” Next, we applied canonical pathway analysis using the IPA library of canonical pathways (part of the Ingenuity Pathways Knowledge Base; IPKB). Eleven canonical pathways were significantly enriched (P < 0.05; Table S1). Of particular interest, several of the significant pathways are related to NAD+/NADH metabolism which has been implicated in neurodegeneration.Citation20,Citation21 Finally, we applied disease association functional analysis, which identifies the diseases that are most significant to the gene set. We found that the top three significantly enriched diseases were “neurological diseases” with enrichment p-value between 1.51E-04 to 5E-02 (Table S2), “psychological disorders” (P = 1.8E to 04–4.98E-02; Table S3), and “skeletal and muscular disorders” (P = 1.8E-04 to 4.77E-02; Table S4) Some of the top neurological diseases were “progressive neuropathy,” “Parkinson's disease,” “attention deficit disorders,” “attention deficit hyperactivity disorder,” “narcolepsy,” and “dementia.” All these findings are consistent with the typical HSAN1E phenotypes which include progressive neuropathy, dementia, sleep disorder, and personality disorders, indicating that the gene set we identified is associated with pathogenesis of central and peripheral neurodegeneration, and may have important implication in other neurodegenerative disorders.
CpG methylation in imprinted genes
Genomic imprinting is an epigenetic regulatory mechanism that turns off expression from either the paternal or the maternal allele to achieve mono-allelic gene expression through DNA methylation and histone modulation. As a DNA methylation maintenance enzyme, DNMT1 has been shown to be the enzyme responsible for maintaining the imprinting process during and after embryogenesis.Citation22,Citation23 Any defect of this enzyme likely leads to reduced methylation in imprinted genes. We analyzed the list of 201 well-characterized imprinted genes obtained from the geneimprint web resource (http://www.geneimprint.org/). Collectively, the CpGs in imprinted genes were more differentially methylated than in non-imprinted genes (5.1% vs. 4.5% CpGs, respectively, Chi-square test P = 9.01 × 10−12). There were more hypomethylated CpGs in the imprinted genes than in other genes (72% vs. 66% hypomethylated CpGs, Chi-square test P = 9.01 × 10−12), suggesting DNMT1 mutation has a higher impact on the imprinted genes.
CpG methylation in histone modification mark sites
To examine whether differential DNA methylation preferentially occurred in regions normally occupied by specific histone marks and variants, we compared the overall CpG methylation between the affected and unaffected siblings in the regions of 11 histone modifications and variants (H2A.Z, H3K4me1, H3K4me2, H3K4me3, H3K9ac, H3K9me3, H3K27ac, H3K27me3, H3K36me3, H3K79me3, H4K20me1) reported in a lymphoblastoid cell line of a healthy donor (GM12878) by the ENCODE project (https://genome.ucsc.edu/ENCODE/). In addition, we also compared the CTCF (CCCTC-Binding factor) binding sites defined in two replicates of the same cell line (CTCF.rep1 and CTCF.rep2). We found that CpG methylation was reduced in the affected individuals in the regions normally occupied by the repressive histone marks H3K27me3 (4.1% median reduction) and H3K9me3 (1.2% median reduction), as well as CTCF (2.3% median reduction) (). Increasing evidence indicates that DNA methylation and histone modification pathways work synergistically to regulate chromatin structure and gene expression. DNA methylation may serve as a template for histone modifications, and decreased methylation in the repressive histone marks may lead to interruption of proper heterochromatin state.
CpG methylation in genes associated with mitochondrial function
Mitochondrial genes perform the important function in energy generation for cells. The fact that HSAN1E patients manifest some clinical presentations similar to mitochondrial diseases or the mutation leads to the loss of high energy demanding neurons prompted us to investigate the methylation profile of mitochondrial genes. We identified 666 mitochondrial function-related genes encoded either in the nuclear or the mitochondrial genome (Table S5), and 654 were captured in the data set with 10× coverage. Comparing the mean methylation of these genes between the affected and unaffected samples, we did not see noticeable difference (0.83 vs. 0.84). About 4.1% of CpGs in genes related to mitochondrial function were differentially methylated vs. 4.5% methylated CpGs found in genes unrelated to mitochondrial function. Of the differentially methylated genes annotated to mitochondrial functions, 54.8% was hypomethylated.
Discussion
Accumulating evidence suggests DNMT1 plays multiple critical functions in the nervous system,Citation24 but the exact molecular mechanism(s) remain unclear. HSAN1E is the first identified adult-onset, monogenic neurodegenerative disease caused by DNMT1 mutations. The disease-causal DNMT1 mutation provides a unique window through which we can investigate specific methylation defects underlying an adult-onset neurodegenerative disorder at the molecular level. The role of DNMT1 is also of fundamental biological interest, as it is the only essential methyltransferase to ensure the fidelity of epigenetic inheritance. Through whole genome methylation evaluation with single base resolution, our study reveals a signature pattern of methylation alteration in this adult onset neurodegenerative disease, characterized by a prevalent hypomethylation in intergenic regions, a significant methylation decrease at surrounding regions of transcription start sites (TSS), and marked methylation reduction on chromosomes X and 18 (5–10%). Additionally, DNMT1 mutation leads to more hypermethylated DMC in some CpG islands.
Our results provide an interesting contrast yet unique resemblance to a recent study that also used whole genome bisulfite sequencing to assess the methylation profile change in a ICF syndrome patient with DNMT3B mutation.Citation25 This study showed DNMT3B mutations cause severe hypomethylation across the whole genome (~25% overall reduction) and profound losses in chromosome X (63%). ICF syndrome patients have autosomal recessive inheritance and an average life expectancy of only 8 y. In contrast, our whole genome bisulfite sequencing results showed that the impact of DNMT1 heterozygous mutation on global genome methylation is only modestly changed in 3 affected HSAN1E patients relative to their age- and gender-matched siblings. However, the strong impact of DNMT1 heterozygous mutation is rather exerted in intergenic regions, TSS regions and chromosomes 18 and X. These regions have between 5–10% reductions in methylation, suggesting a targeted and insidious effect of DNMT1 methylation defect. HSAN1E patients have autosomal dominant inheritance; they are normal from birth to early teenager years, but then start to develop progressive sensory neuropathy, sensorineural hearing loss and memory loss in young adulthood. The life expectancy of HSAN1E is approximately 50 y and the cause of death is usually related to dementia complications. The ICF syndrome’s early-onset and severe phenotypes are consistent with the drastic methylation decrease while HSAN1E’s adult-onset feature, unique phenotypes and progressive course are linked with region-specific methylation reduction. Our findings suggest DNMT1 heterozygous mutation has minimal effect during development, but has deleterious accumulating impact on the nervous system over a long period of time and specifically on memory formation and sensory neurons.
Because patients with DNMT1 mutations have a syndromic appearance similar to many mitochondrial disorders, we investigated the DMC pattern of mitochondrial function-related genes encoded either in the nuclear or the mitochondrial genome, but did not find significant difference. However, our IPA analysis demonstrated that the 1693 genes associated with DMRs are highly enriched in many neurological and psychological disorders. Alzheimer disease, narcolepsy, and dementia were among the top significantly associated diseases. The canonical pathway analysis revealed NAD+/NADH metabolism pathways are implicated in the pathogenesis. NAD+ is a coenzyme involved in redox reactions, expressed ubiquitously and has critical functions in the neural cells. NAD+ has been shown not only important in energy metabolism and mitochondrial functions, but also in calcium homeostasis, aging, and cell death. In animal models, increased nuclear NAD+ has neuroprotective function against the negative effects of both mechanical and chemical insult.Citation26 Emerging evidence shows NAD+ as an important therapeutic target for the treatment of age-related degenerative diseases, such as Alzheimer disease.Citation27,Citation28 Our result indicates that DNMT1 likely impacts a specific set of pathway involving NAD+/NADH metabolism that are particularly imperative in degenerative neurological disorders.
Increasingly epigenetic mechanisms are also being recognized important in sporadic neurological diseases without apparent genetic causes.Citation5,Citation29 The high fidelity DNMT1 enzyme activity is critical in epigenetic regulation. Our DNMT1 Mendelian disease model provides a unique and valuable avenue for future research in common varieties of human neurodegenerative disorders. Utilizing a unique paired-sample design, our study showed that even modest reductions in DNMT1 enzyme activity and preferential site-specific intergenic changes of DNA methylation can lead to devastating neurological consequences. Understanding how DNMT1 mutation exerts its deleterious impact in a region- and chromosome- specific manner and how the methylation changes specifically lead to sensory nerve axonal loss and brain atrophy with cognitive decline will require a significant amount of future study. Our work provided first insights into a specific neurodegenerative pathway by identifying a signature genome methylation pattern potentially having broader implications beyond HSAN 1E.
Materials and Methods
Study subjects
Since methylation is associated with age and gender, we identified three same-gender, similar aged (<5 y) sibling pairs from the large kindred of European origins (Caucasian) we longitudinally studied in our original publication for whole genome methylation analysis. Three HSAN1patients with DNMT1 heterozygous Y495C mutation were matched with their normal siblings without the mutation. Their genomic DNA samples were collected from blood and whole genome bisulfite sequencing was performed. The clinical features are very similar between the affected persons in this kindred.Citation8,Citation30,Citation31 They all started to lose their sensory nerve action potentials during young adulthood. By the 3rd-4th decade, prominent sensorineural hearing loss with cognitive and behavioral changes was noted with progressive declines, leading to the requirement of total care, and death often occurred in the 4th-5th decades. Brain imaging and limited autopsy of the affected persons showed global atrophy and a reduced weight of the autopsied brains, spinal cord posterior column loss of axons, and pansensory fiber loss in distal nerves. Histopathologic analysis did not reveal any inclusion bodies such as β-amyloid, tau, TDP-43, and α-synuclein.Citation8,Citation30 The study protocols were approved by Mayo Clinic Institute Review Boards.
DNA extraction
DNA samples were extracted from patients’ blood. Briefly, the anticoagulated blood samples were centrifuged at 2500 g for 10 min, and the separated buffy coat layers were collected. Then Genome DNA was extract from buffy coat using Qiamp DNA Blood Kit (Qiagen).
Whole genome bisulfite methylation sequencing
10μg genomic DNA was used for bisulfite sequencing library construction according to the standard Illumina Paired-End protocol. Briefly, genomic DNA was fragmented and adaptors were ligated. DNA samples were then subjected to the bisulfite conversion using EZ DNA Methylation-Gold kit (Zymo), purified using AMPure beads (Agencourt) and amplified by 10 cycles PCR. Sequencing was performed on HiSeq2000 (Illumina) with single-end reads on HiSeq2000 using TruSeq SBS sequencing kit version 3 and HCS v2.0.12 data collection software.
WGBS data analysis
The raw sequence fastq data were processed using our bisulfite sequence analytical pipeline, SAAP-BS (v1)Citation32 with alignment to human reference genome version 19 (hg19) to obtain CpG site methylation including both methylated and unmethylated cytosine counts and methylation ratio. All CpG sites were extracted. To ensure data quality and reliability, we only included CpG sites with ≥10× coverage in the downstream differential methylation CpG analyses. Overall methylation was measured by mean and median genome wide, by chromosome, or special regions of the genome for each individual or comparison group. DMC detection between affected and unaffected siblings was performed by paired t statistics. The constant CpGs across samples (the methylation ratio is either = 1 or = 0 in all six samples) were first removed. CpGs with P < 0.05 in paired t test were considered differentially methylated. The distribution of these DMCs in the genome was summarized against various genomic regions including intergenic, genic regions, promoters, repetitive sequences, histone modification, and imprinted genes.
Differential methylation region analysis
Differentially methylated regions (DMRs) between affected and unaffected siblings were identified using the R package “bsseq” as previously described.Citation18 For this detection analysis, we retained all CpG sites that were sequenced in all six samples regardless of coverage. A smoothing approach was used to stabilize the sites with a lower coverage since nearby CpGs tend to have similar methylation patterns.Citation33 The smoothing procedures regresses these CpGs to reduce potential outliers at low-covered CpG sites. T statistics was then performed for each CpG independently and similarly changed CpGs were grouped into DMRs according to specified minimum CpG number and methylation difference between affected and unaffected individuals (minimum of 5 CpGs and at least 10% methylation difference in the DMR region were used).
Pathway/gene enrichment analyses by ingenuity pathway analysis
For the genes with DMRs within 5kb of TSS, we conducted a pathway, gene network and functional enrichment analysis using IPA (http://www.ingenuity.com/, performed on September 5, 2013), a widely used pathway and system biology interpretation toolCitation19 in order to identify relevant biological and molecular networks. IPA uses a proprietary database created from continuously updated, published, peer-reviewed scientific publications stored in the Ingenuity Pathways Knowledge Base (IPKB). It offers several function modules including Core, Tox, Metabolomics, Comparison, Biomarker Filter, and microRNA Target Analysis. The Core Analysis was used for this data, which includes the enrichment for canonical pathway, disease/biological function, toxicological function and gene-gene interaction networks. The enrichment P value is calculated using the right-tailed Fisher Exact Test, representing the likelihood of the association between a set of focus genes and a given process or pathway is due to random chance.
| Abbreviations: | ||
| WGBS | = | whole genome bisulfite sequencing |
| HSAN1E | = | Hereditary Sensory and Autonomic Neuropathy 1E |
| DMCs | = | differentially methylated CpGs |
| DMRs | = | differentially methylated regions |
| CGI | = | CpG island |
Additional material
Download Zip (260.1 KB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported in part by the Epigenomics Translational Program and the Bioinformatics Program of the Mayo Clinic Center for Individualized Medicine and the National Institutes of Health (K08 NS065007).
References
- Qureshi IA, Mehler MF. Epigenetic mechanisms governing the process of neurodegeneration. Mol Aspects Med 2013; 34:875 - 82; http://dx.doi.org/10.1016/j.mam.2012.06.011; PMID: 22782013
- Bergman Y, Cedar H. DNA methylation dynamics in health and disease. Nat Struct Mol Biol 2013; 20:274 - 81; http://dx.doi.org/10.1038/nsmb.2518; PMID: 23463312
- Jair KW, Bachman KE, Suzuki H, Ting AH, Rhee I, Yen RW, Baylin SB, Schuebel KE. De novo CpG island methylation in human cancer cells. Cancer Res 2006; 66:682 - 92; http://dx.doi.org/10.1158/0008-5472.CAN-05-1980; PMID: 16423997
- Liang G, Chan MF, Tomigahara Y, Tsai YC, Gonzales FA, Li E, Laird PW, Jones PA. Cooperativity between DNA methyltransferases in the maintenance methylation of repetitive elements. Mol Cell Biol 2002; 22:480 - 91; http://dx.doi.org/10.1128/MCB.22.2.480-491.2002; PMID: 11756544
- Marques SC, Oliveira CR, Pereira CM, Outeiro TF. Epigenetics in neurodegeneration: a new layer of complexity. Prog Neuropsychopharmacol Biol Psychiatry 2011; 35:348 - 55; http://dx.doi.org/10.1016/j.pnpbp.2010.08.008; PMID: 20736041
- Klein CJ, Benarroch EE. Epigenetic regulation: basic concepts and relevance to neurologic disease. Neurology 2014; 82:1833 - 40; http://dx.doi.org/10.1212/WNL.0000000000000440; PMID: 24759839
- Xu GL, Bestor TH, Bourc’his D, Hsieh CL, Tommerup N, Bugge M, Hulten M, Qu X, Russo JJ, Viegas-Péquignot E. Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature 1999; 402:187 - 91; http://dx.doi.org/10.1038/46214; PMID: 10647011
- Klein CJ, Botuyan MV, Wu Y, Ward CJ, Nicholson GA, Hammans S, Hojo K, Yamanishi H, Karpf AR, Wallace DC, et al. Mutations in DNMT1 cause hereditary sensory neuropathy with dementia and hearing loss. Nat Genet 2011; 43:595 - 600; http://dx.doi.org/10.1038/ng.830; PMID: 21532572
- Winkelmann J, Lin L, Schormair B, Kornum BR, Faraco J, Plazzi G, Melberg A, Cornelio F, Urban AE, Pizza F, et al. Mutations in DNMT1 cause autosomal dominant cerebellar ataxia, deafness and narcolepsy. Hum Mol Genet 2012; 21:2205 - 10; http://dx.doi.org/10.1093/hmg/dds035; PMID: 22328086
- Moghadam KK, Pizza F, La Morgia C, Franceschini C, Tonon C, Lodi R, Barboni P, Seri M, Ferrari S, Liguori R, et al. Narcolepsy is a common phenotype in HSAN IE and ADCA-DN. Brain 2014; 137:1643 - 55; http://dx.doi.org/10.1093/brain/awu069; PMID: 24727570
- Klein CJ, Bird T, Ertekin-Taner N, Lincoln S, Hjorth R, Wu Y, Kwok J, Mer G, Dyck PJ, Nicholson GA. DNMT1 mutation hot spot causes varied phenotypes of HSAN1 with dementia and hearing loss. Neurology 2013; 80:824 - 8; http://dx.doi.org/10.1212/WNL.0b013e318284076d; PMID: 23365052
- Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 1992; 69:915 - 26; http://dx.doi.org/10.1016/0092-8674(92)90611-F; PMID: 1606615
- Chen T, Hevi S, Gay F, Tsujimoto N, He T, Zhang B, Ueda Y, Li E. Complete inactivation of DNMT1 leads to mitotic catastrophe in human cancer cells. Nat Genet 2007; 39:391 - 6; http://dx.doi.org/10.1038/ng1982; PMID: 17322882
- Ma DK, Guo JU, Ming GL, Song H. DNA excision repair proteins and Gadd45 as molecular players for active DNA demethylation. Cell Cycle 2009; 8:1526 - 31; http://dx.doi.org/10.4161/cc.8.10.8500; PMID: 19377292
- Klein CJ. DNMT1-Related Dementia, Deafness, and Sensory Neuropathy. In: Pagon RA, Bird TD, Dolan CR, Stephens K, Adam MP, eds. GeneReviews. Seattle (WA), 1993.
- Ehrlich M, Gama-Sosa MA, Huang LH, Midgett RM, Kuo KC, McCune RA, Gehrke C. Amount and distribution of 5-methylcytosine in human DNA from different types of tissues of cells. Nucleic Acids Res 1982; 10:2709 - 21; http://dx.doi.org/10.1093/nar/10.8.2709; PMID: 7079182
- Bird A, Taggart M, Frommer M, Miller OJ, Macleod D. A fraction of the mouse genome that is derived from islands of nonmethylated, CpG-rich DNA. Cell 1985; 40:91 - 9; http://dx.doi.org/10.1016/0092-8674(85)90312-5; PMID: 2981636
- Hansen M, Gerds TA, Nielsen OH, Seidelin JB, Troelsen JT, Olsen J. pcaGoPromoter--an R package for biological and regulatory interpretation of principal components in genome-wide gene expression data. PLoS One 2012; 7:e32394; http://dx.doi.org/10.1371/journal.pone.0032394; PMID: 22384239
- Thomas S, Bonchev D. A survey of current software for network analysis in molecular biology. Hum Genomics 2010; 4:353 - 60; http://dx.doi.org/10.1186/1479-7364-4-5-353; PMID: 20650822
- Araki T, Sasaki Y, Milbrandt J. Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science 2004; 305:1010 - 3; http://dx.doi.org/10.1126/science.1098014; PMID: 15310905
- Raff MC, Whitmore AV, Finn JT. Axonal self-destruction and neurodegeneration. Science 2002; 296:868 - 71; http://dx.doi.org/10.1126/science.1068613; PMID: 11988563
- Hirasawa R, Chiba H, Kaneda M, Tajima S, Li E, Jaenisch R, Sasaki H. Maternal and zygotic Dnmt1 are necessary and sufficient for the maintenance of DNA methylation imprints during preimplantation development. Genes Dev 2008; 22:1607 - 16; http://dx.doi.org/10.1101/gad.1667008; PMID: 18559477
- Smith ZD, Chan MM, Mikkelsen TS, Gu H, Gnirke A, Regev A, Meissner A. A unique regulatory phase of DNA methylation in the early mammalian embryo. Nature 2012; 484:339 - 44; http://dx.doi.org/10.1038/nature10960; PMID: 22456710
- Lister R, Mukamel EA, Nery JR, Urich M, Puddifoot CA, Johnson ND, Lucero J, Huang Y, Dwork AJ, Schultz MD, et al. Global epigenomic reconfiguration during mammalian brain development. Science 2013; 341:1237905; http://dx.doi.org/10.1126/science.1237905; PMID: 23828890
- Heyn H, Vidal E, Sayols S, Sanchez-Mut JV, Moran S, Medina I, Sandoval J, Simó-Riudalbas L, Szczesna K, Huertas D, et al. Whole-genome bisulfite DNA sequencing of a DNMT3B mutant patient. Epigenetics 2012; 7:542 - 50; http://dx.doi.org/10.4161/epi.20523; PMID: 22595875
- Bedalov A, Simon JA. Neuroscience. NAD to the rescue. Science 2004; 305:954 - 5; http://dx.doi.org/10.1126/science.1102497; PMID: 15310883
- Belenky P, Racette FG, Bogan KL, McClure JM, Smith JS, Brenner C. Nicotinamide riboside promotes Sir2 silencing and extends lifespan via Nrk and Urh1/Pnp1/Meu1 pathways to NAD+. Cell 2007; 129:473 - 84; http://dx.doi.org/10.1016/j.cell.2007.03.024; PMID: 17482543
- Belenky P, Bogan KL, Brenner C. NAD+ metabolism in health and disease. Trends Biochem Sci 2007; 32:12 - 9; http://dx.doi.org/10.1016/j.tibs.2006.11.006; PMID: 17161604
- Qureshi IA, Mehler MF. Advances in epigenetics and epigenomics for neurodegenerative diseases. Curr Neurol Neurosci Rep 2011; 11:464 - 73; http://dx.doi.org/10.1007/s11910-011-0210-2; PMID: 21671162
- Hojo K, Imamura T, Takanashi M, Ishii K, Sasaki M, Imura S, Ozono R, Takatsuki Y, Takauchi S, Mori E. Hereditary sensory neuropathy with deafness and dementia: a clinical and neuroimaging study. Eur J Neurol 1999; 6:357 - 61; http://dx.doi.org/10.1046/j.1468-1331.1999.630357.x; PMID: 10210919
- Wright A, Dyck PJ. Hereditary sensory neuropathy with sensorineural deafness and early-onset dementia. Neurology 1995; 45:560 - 2; http://dx.doi.org/10.1212/WNL.45.3.560; PMID: 7898717
- Sun Z, Baheti S, Middha S, Kanwar R, Zhang Y, Li X, Beutler AS, Klee E, Asmann YW, Thompson EA, et al. SAAP-RRBS: streamlined analysis and annotation pipeline for reduced representation bisulfite sequencing. Bioinformatics 2012; 28:2180 - 1; http://dx.doi.org/10.1093/bioinformatics/bts337; PMID: 22689387
- Bird A. DNA methylation patterns and epigenetic memory. Genes Dev 2002; 16:6 - 21; http://dx.doi.org/10.1101/gad.947102; PMID: 11782440