
Abstract
Changes to the DNA methylome have been described in patients with rheumatoid arthritis (RA). In previous work, we reported genome-wide methylation differences in T-lymphocyte and B-lymphocyte populations from healthy individuals. Now, using HumanMethylation450 BeadChips to interrogate genome-wide DNA methylation, we have determined disease-associated methylation changes in blood-derived T- and B-lymphocyte populations from 12 female patients with seropositive established RA, relative to 12 matched healthy individuals. Array data were analyzed using NIMBL software and bisulfite pyrosequencing was used to validate array candidates. Genome-wide DNA methylation, determined by analysis of LINE-1 sequences, revealed higher methylation in B-lymphocytes compared with T-lymphocytes (P ≤ 0.01), which is consistent with our findings in healthy individuals. Moreover, loci-specific methylation differences that distinguished T-lymphocytes from B-lymphocytes in healthy individuals were also apparent in RA patients. However, disease-associated methylation differences were also identified in RA. In these cases, we identified 509 and 252 CpGs in RA-derived T- and B-lymphocytes, respectively, that showed significant changes in methylation compared with their cognate healthy counterparts. Moreover, this included a restricted set of 32 CpGs in T-lymphocytes and 20 CpGs in B-lymphocytes (representing 15 and 10 genes, respectively, and including two, MGMT and CCS, that were common to both cell types) that displayed more substantial changes in methylation. These changes, apparent as hyper- or hypo-methylation, were independently confirmed by pyrosequencing analysis. Validation by pyrosequencing also revealed additional sites in some candidate genes that also displayed altered methylation in RA. In this first study of genome-wide DNA methylation in individual T- and B-lymphocyte populations in RA patients, we report disease-associated methylation changes that are distinct to each cell type and which support a role for discrete epigenetic regulation in this disease.
Introduction
Rheumatoid arthritis (RA) is a systemic autoimmune disease of largely unknown cause that manifests predominantly as chronic inflammation of the synovial joints, leading to progressive joint destruction and functional impairment. It is widely believed that a complex interplay between genetic factors and the environment is integral to the disease process. Epigenetic mechanisms, including DNA methylation, histone modifications and expression of microRNAs (miRNAs), impact on gene expression without changing the underlying DNA sequence itself.Citation1 These changes, frequently acting in concert, are thought to represent the likely intermediaries through which the environment can impact on disease, including autoimmune rheumatic diseases.Citation2 Indeed, an expanding literature now describes changes to the epigenetic landscape in patients with RA and related conditions (reviewed in refs. Citation3, Citation4).
In RA, distinct leukocyte populations, including T- and B-lymphocytes and their subsets, are key players driving autoimmunity and chronic inflammation. The development and differentiation of the different T- and B-lymphocyte subsets/lineages is tightly regulated, and epigenetic mechanisms including DNA methylation appear to play an essential role in this process.Citation5,Citation6 Methylation of the DNA base cytosine in the context of promoter-associated cytosine-guanine dinucleotides (CpGs) is frequently associated with transcriptional repression and gene silencing.Citation7 In this context, DNA methylation in T-lymphocytes is known to play an essential role in lineage commitment to the T helper 1 (Th1) and Th2 subsets through regulation of interferon-γ and interleukin-4 expressionCitation8,Citation9 and the expression of the transcription factors STAT4 and STAT6.Citation10,Citation11 Similarly, development of regulatory T-lymphocytes (Treg), essential for immunological self-tolerance,Citation12 is epigenetically controlled, in this case mediated through the methylation status of the locus encoding the FOXP3 transcription factor that drives Treg differentiation.Citation13
Several studies have reported altered levels of global DNA methylation in lymphocytes and peripheral blood mononuclear cells from patients with RA.Citation14,Citation15 Moreover, candidate gene studies in RA describe DNA methylation changes in these cells for a number of different genes, including interleukin-6 (IL6),Citation16 IL10,Citation17 EFNB1,Citation18 and CD40LG.Citation19 In patients with other rheumatological conditions, including systemic lupus erythematosus and juvenile idiopathic arthritis, evidence from genome-wide studies indicates distinct methylation changes in multiple genes in T-lymphocytes.Citation20,Citation21 To date, however, no systematic genome-wide analysis of DNA methylation in individual T- and B-lymphocyte populations has been described in patients with RA. Indeed, to our knowledge, we were the first to recently characterize intrinsic site-specific differences in genome-wide DNA methylation between matched pairs of T- and B-lymphocytes from healthy individuals.Citation22
These observations strongly support the need to interrogate DNA methylation in individual lymphocyte populations that drive disease in RA, an approach that will help establish disease-associated and cell type-specific methylation changes in these patients. Following our recent work examining T- and B-lymphocytes from healthy individuals,Citation22 we now describe genome-wide methylation profiles in T-lymphocyte and B-lymphocyte populations isolated from patients with RA.
Results
Technical validation of array data
High-resolution genome-wide arrays (HumanMethylation450 BeadChip, Illumina Inc.; henceforth referred to as ‘array’ or ‘450k array’) were used to quantify DNA methylation at over 480,000 unique CpG sites across the genome in T- and B-lymphocyte populations collected from a cohort of RA patients and healthy individuals. A technical validation between array β-values and methylation levels as determined by bisulfite pyrosequencing revealed a strong positive correlation (Spearman’s r = 0.889, P < 0.00001; Fig. S1). However, as in our previous study,Citation22 and as reported by others,Citation23 pyrosequencing analyses generally reported lower methylation levels than the respective array β-values, in particular across the mid-range of methylation β-values (30–70%).
Global DNA methylation in RA derived T- and B-lymphocytes
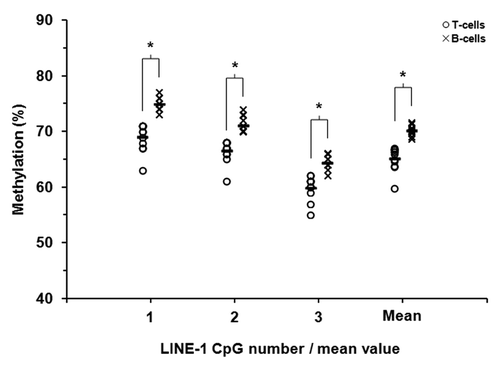
Our previous report in healthy individuals revealed a subtle yet statistically significant increase in LINE-1 DNA methylation in B-lymphocytes compared with T-lymphocytes.Citation22 We therefore determined, for this surrogate measure of global methylation, the relationship between these two cell types in patients with RA. As in our previous report, examination of three CpG dinucleotides in the LINE-1 sequence revealed a modest but statistically significant increase in mean global methylation in B-lymphocytes relative to patient matched T-lymphocytes (70.2 vs. 65.2%, P = 0.0001, Wilcoxon Signed-Rank Test) (). The observed increase was evident for each of the individual sites examined (P < 0.0005 in each case). Assessment of global methylation using a more gene-focused approach by inspecting the average β-value across all array CpGs revealed no significant difference between the cell types, as we had noted in healthy individuals (β = 0.566 and 0.558 for T- and B-lymphocytes, respectively, P > 0.1). Global DNA methylation in RA patients therefore appears to closely reflect our observations in healthy individuals.
Figure 1. LINE-1 DNA methylation in T-lymphocytes and B-lymphocytes from patients with rheumatoid arthritis. Methylation at three adjacent CpG sites within LINE-1 repetitive sequences was quantified in purified T- and B-lymphocytes from 12 RA patients by sodium bisulfite pyrosequencing. Unfilled circles and crosses represent T-lymphocytes and B-lymphocytes, respectively. The mean levels of methylation for each of the CpG sites are shown by the short black horizontal bars.* P ≤ 0.01 (Wilcoxon Signed-Rank Test).
Cell type-specific DNA methylation in RA derived T- and B-lymphocytes
Through interrogation of DNA methylation in purified T- and B-lymphocyte populations in healthy individuals, we recently identified 679 individual CpG sites that defined a unique methylation profile that distinguishes between and defines these two lymphocyte types.Citation22 Hierarchical clustering across these 679 sites in T- and B-lymphocytes from RA patients revealed an essentially identical methylation profile, where samples segregated into two distinct clusters based on cell type (; the reader is also directed to our previous publication for comparisonCitation22). No sites additional to those described above showed methylation differences between the cell types that were unique to RA patients. Overall, these data indicate that intrinsic methylation differences that distinguish T-lymphocytes from B-lymphocytes are recapitulated in RA patients.
Figure 2. DNA methylation heatmap for the 679 CpG methylation signature in RA derived T- and B-lymphocytes. CpGs presented in the heatmap were described in our previous work (22) and represent those sites which define a unique methylation signature distinguishing healthy T-lymphocytes from B-lymphocytes. Each row represents an individual CpG, ordered according to the clustering output in healthy individuals (22), and each column a different sample (listed beneath the heatmap). Color gradation from yellow to blue represents low to high DNA methylation respectively, with β-values ranging from 0 (no methylation; yellow) to 1 (complete methylation; blue). Intrinsic methylation differences previously observed between the cell types in healthy individuals are accurately preserved in RA patients.

T- and B-lymphocytes from RA patients show altered DNA methylation for a restricted and distinct set of CpGs
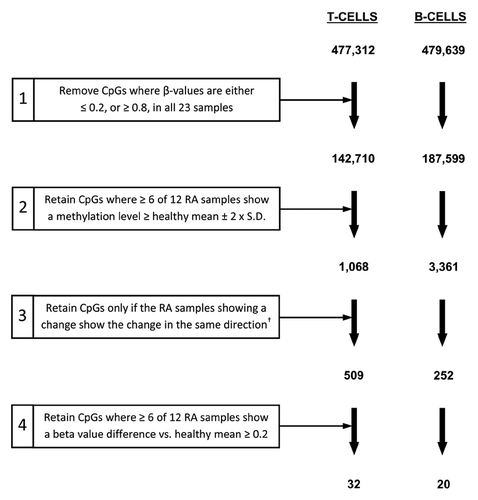
To identify potential disease-associated methylation changes in RA derived T- and B-lymphocytes, we next compared each of the cell types with their cognate healthy cell counterparts. In this case, we used a series of criteria to filter CpGs in each data set, retaining only those meeting each successive step (). Using an approach that we and others have previously taken,Citation22,Citation24 we first removed all non-variable CpG sites from the data set (sites for which methylation β-values in all 23 samples were ≤ 0.2, or ≥ 0.8). We then directly compared DNA methylation in RA derived T- and B-lymphocytes with their cognate (T or B) counterpart sample from healthy individuals. To account for the variation in β-values observed at individual sites within the RA group, we only considered a CpG to be differentially methylated in this group if six or more of the 12 patients demonstrated a methylation β-value that differed from the mean of the healthy group by two or more times (plus or minus) the standard deviation (step 2, ). This approach is similar to one we have used previously to identify disease-associated differentially methylated CpGs.Citation25 By retaining only those sites showing methylation changes in the same direction (hypermethylation or hypomethylation in individual RA samples; step 3, ), we identified 509 and 252 CpGs displaying disease-associated methylation differences in RA derived T- and B-lymphocytes, respectively. Moreover, this included a restricted set of 32 and 20 CpGs in T- and B-lymphocytes which displayed larger differences in methylation β-value that were at least 0.2 greater/lower than the average observed in their cognate healthy counterparts and that were distinct to either cell type.
Figure 3. Filtering criteria for identification of CpGs differentially methylated between RA patients and healthy individuals. For T-lymphocytes and B-lymphocytes separately, the starting number of CpGs (477,312 and 479,639 for T- and B-lymphocytes respectively) was derived through the removal of unreliable sites (CpGs with detection p-values > 0.05) and those with missing β-values, as described in the Methods. Numbers indicate the number of CpGs remaining at each step.† Refers to CpGs for which all RA samples showing a change from the healthy mean ± two-times the SD in the preceding step were either hypermethylated or hypomethylated in RA.
Summary data for the 32 and 20 CpGs identified according to the criteria described above as differentially methylated in T- and B-lymphocytes respectively are presented in (a complete list and annotation for the CpGs/genes is provided in Table S1). Of the 32 CpGs identified in T-lymphocytes, approximately equal numbers were hypermethylated as were hypomethylated. Conversely, all 20 CpGs identified in B-lymphocytes were hypomethylated in RA relative to their corresponding healthy counterparts. In both cell types approximately half of the CpGs identified were associated with known genes (18/32 and 10/20, T- and B-lymphocytes respectively). The majority of CpGs identified in T-lymphocytes were located within a CpG island or the surrounding island shores and shelves (19; 59.4%). In contrast, fewer than half (8/20) of the CpGs identified in B-lymphocytes were associated with a CpG island or surrounding regions. Four of the candidate sites identified were common to both cell types, of which two were associated with known genes (MGMT [O-6-methylguanine-DNA methyltransferase] and CCS [Copper chaperone for superoxide dismutase]).
Table 1. Summary characteristics for the differentially methylated CpGs identified in RA derived T-lymphocytes and B-lymphocytes.*
Hierarchical clustering based on the 32 and 20 candidate CpGs identified in T- and B-lymphocytes respectively was suggestive of patients and healthy controls clustering independently, although perfect segregation of the two groups was not observed (Fig. S2). While this may reflect the absolute numbers used in the analysis, an interesting observation was the identification of a cluster of six RA patients where methylation across the 32 CpG candidates in T-lymphocytes was distinct from the other six patients and healthy controls. However, examination of known clinical characteristics in these patients, including measures of systemic inflammation, disease activity and functional outcome, did not reveal any differences relative to these measures in the other six patients.
Independent validation of candidate CpGs
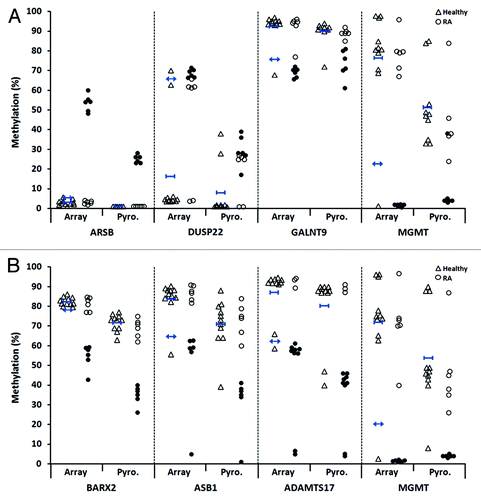
To confirm the altered methylation of candidate CpGs identified in our genome-wide approach, we selected eight individual sites, four of these from the T-lymphocyte candidates (ARSB, DUSP22, GALNT9 and MGMT) and four from the B-lymphocyte candidates (ADAMTS17, ASB1, BARX2 and MGMT), for independent validation by bisulfite pyrosequencing. We specifically focused on CpGs that were associated with a gene and/or CpG island and its surrounding region (assay details are provided in Table S2). As presented in , the individual RA samples that were identified as differentially methylated for each of the candidate sites by array analysis (filled circles) were confirmed by pyrosequencing. In each case, the same individual RA samples showed methylation changes consistent with those at the array-interrogated site (filled circles in the pyrosequencing data indicate the same individual samples denoted by filled circles in the array data).
Figure 4. Validation by bisulfite pyrosequencing of differentially methylated CpG candidates in RA derived T- and B-lymphocytes. For each cell type, four candidate CpGs that were differentially methylated between RA patients and healthy individuals were selected. This included two sites each that were hypermethylated (ARSB and DUSP22) and hypomethylated (GALNT9 and MGMT) in RA derived T-lymphocytes (A), and included four hypomethylated sites in B-lymphocytes (B). In each plot, unfilled triangles indicate healthy individuals and where the mean methylation and two-times the SD of the mean are indicated for the array data by short blue horizontal bars with vertical stops and with arrowheads, respectively. RA patients are indicated by circles, where filled circles in the array data indicate the individual samples that were identified as differentially methylated relative to healthy individuals (as defined by the criteria in ). These same samples are also indicated by filled circles in the pyrosequencing data. Abbreviations: Pyro., bisulfite pyrosequencing.
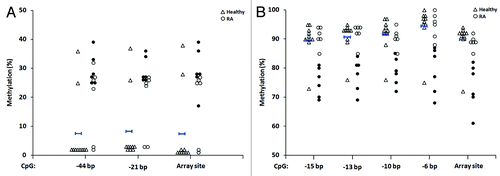
Pyrosequencing analysis also revealed that for both DUSP22 (dual specificity phosphatase 22; two sites) and GALNT9 (UDP-N-acetyl-α-D-galactosamine:polypeptide N-acetylgalactosaminyltransferase 9; four sites), additional CpGs juxtaposed to the array-identified site also showed methylation changes in RA patients that were consistent with those observed at the array site (hypermethylation for DUSP22 and hypomethylation for GALNT9; ). In addition to the validated candidates described, we also sought to examine by pyrosequencing several other array-identified candidate CpGs, but in these cases noted the presence of single nucleotide polymorphisms at the interrogated site. Validation of multiple candidates reinforces the robustness of the array data and our approach to identify sites differentially methylated in T- and B-lymphocytes derived from patients with RA.
Figure 5. Identification of additional differentially methylated CpGs for two array-identified candidates in RA derived T-lymphocytes. Shown are additional CpGs adjacent to the array-identified candidate site for DUSP22 (two additional sites) (A) and GALNT9 (four additional sites) (B) that were also found to be differentially methylated in RA patients relative to healthy individuals, as determined by pyrosequencing. Negative base pair numbers on the x-axis indicate bases upstream of the array CpG site. In each plot, unfilled triangles indicate healthy individuals and where the mean methylation is indicated by short blue horizontal bars with vertical stops. RA patients are indicated by circles, where filled circles for the array site indicate the individual samples that were identified as differentially methylated relative to healthy individuals (as defined by the criteria in ). These same samples are also indicated by filled circles in each of the adjacent CpGs presented. Abbreviations: bp, base pairs.
Discussion
Changes to the epigenetic landscape, as defined by DNA methylation, histone modifications and the expression of miRNAs, are apparent in multiple human diseases. Indeed these changes encompass a broad spectrum of autoimmune disorders such as RA, a disease in which lymphocytes and other immune cells play an essential role in autoimmunity and chronic inflammation. In RA, investigations of DNA methylation have typically analyzed global methylcytosine levels or have adopted candidate gene approaches to examine synovial joint cells or cells isolated from blood.Citation14-Citation19,Citation26,Citation27 More recently, several groups have used genome-wide arrays to examine DNA methylation in RA synovial fibroblastsCitation28,Citation29 or the mononuclear cell fraction extracted from whole blood.Citation30 However, and to our knowledge, no array-based studies have examined individual lymphocyte populations. We now report a systematic genome-wide interrogation of DNA methylation in highly purified T- and B-lymphocyte populations from patients with RA. Through comparison with methylation levels determined in their equivalent healthy counterparts we have identified and validated a restricted set of CpGs that show distinct methylation differences in T- and B-lymphocytes from patients with RA.
Our investigation of individual lymphocyte populations in patients with RA is an extension of previous studies in which we specifically sought to define intrinsic methylation differences between highly purified T- and B-lymphocyte populations derived from healthy individuals.Citation22 For those cell populations we described a unique methylation profile that was consistent across individuals and which distinguished the two cell types from each other. This enabled us to investigate DNA methylation in these cells derived from RA patients with a priori knowledge of the differences defining each lymphocyte type. Strikingly, and as illustrated by clustering and heatmap analysis, we found that an almost identical methylation profile to that we initially reported in healthy individualsCitation22 was apparent in purified T- and B-lymphocyte populations from RA patients. However, we did not identify any further changes in methylation between these two cell populations (T-lymphocytes relative to B lymphocytes) that were unique to RA patients i.e. disease-associated. This is supported by our additional finding that methylation within LINE-1 sequences, as a surrogate measure for global methylation, was no different between patients and healthy individuals, for either cell type. The differences observed between the two cell types in RA therefore reflect methylation differences that are intrinsic to the cellular phenotypes (rather than those that are associated with the disease). Similarly, work from other groups has demonstrated that the major blood-derived leukocyte populations show broad-scale differences in methylation in healthy individuals.Citation31
T- and B-lymphocytes are known to play an important role in autoimmunity and the chronic inflammation associated with RA, including, for example, the production of inflammatory cytokines and other mediators, reciprocal interactions with immune and synovial cells, and in the case of B-lymphocytes, antigen presentation and release of autoantibodies [reviewed in refs. Citation32, Citation33]. Through the analysis of these specific cell populations in RA patients and healthy individuals, the principle finding of our study was the identification and subsequent validation of methylation differences at multiple individual CpG sites that were disease-associated and that were distinct to either the T- or B-lymphocyte populations relative to their corresponding healthy counterparts (32 and 20 CpGs in each cell type, respectively). These sites represented 15 genes in T-lymphocytes and 10 genes in B-lymphocytes, and included two genes that were common to both cell types (MGMT and CCS).
Among the CpGs identified as differentially methylated in RA derived T-lymphocytes, six sites within the gene coding for dual specificity phosphatase 22 (DUSP22) (four identified by array and a further two by pyrosequencing) were found to be hypermethylated in RA patients. These sites demonstrated increased methylation in the majority of patients and were associated with a CpG island/shore. DUSP22 is a protein tyrosine phosphatase that, among other actions, has been shown to negatively regulate STAT3 and the IL-6/STAT3-mediated signaling pathway.Citation34 IL-6 is a pivotal cytokine in RA which contributes to autoimmunity, chronic inflammation and joint destruction and the inhibition of which is an effective treatment in this disease.Citation35 The expression and activation of STAT3 in synovial tissues and circulating lymphocytes is also elevated in RA patients.Citation36-Citation38 A recent study has also described a STAT3-inducible gene expression signature in T-lymphocytes from patients with RA.Citation39 Interestingly, altered methylation of DUSP22 has recently been reported in CD4+ T-lymphocytes from patients with the autoimmune condition Sjögren’s syndrome.Citation40 Our data suggest a possible impact of epigenetic alterations in DUSP22 that may influence its role as a negative regulator of the IL-6/STAT3-mediated signaling pathway. We also identified, through the use of array and pyrosequencing analysis, multiple sites within GALNT9 that were consistently hypomethylated in T-lymphocytes from RA patients. The protein product of this gene is one of several glycosyltransferases that are essential in the first step of mucin biosynthesis,Citation41 large glycosylated proteins that have been implicated in RA pathogenesis.Citation42,Citation43 We recognize however, that further investigations of these and other candidates in independent patient cohorts is warranted.
Although a number of different genes have been reported to show methylation changes in RA derived leukocytes,Citation16-Citation19 to our knowledge we are the first to examine and report DNA methylation changes in RA derived B-lymphocytes. In these cells, all of the differentially methylated CpGs we identified were hypomethylated in six or more of the RA patients and relative to their equivalent healthy counterparts. Interestingly, of the 10 sites that were associated with specific genes, seven were located within the gene body (intragenic), a region in which DNA methylation shows positive correlation with transcription levels in B-lymphocytes and other cell types.Citation44,Citation45 In a similar manner, the majority of gene-associated hypomethylated sites we identified in RA derived T-lymphocytes were also located within the gene body. However, and in contrast to these findings, nearly all of the sites identified as hypermethylated in RA patients (15 of 17; all in T-lymphocytes) were located either within a CpG island – regions of CpG-rich DNA that co-localize with 40–60% of gene promotersCitation46,Citation47 – or, more frequently, within the regions surrounding these islands, termed CpG island shores and shelves, which may be important in the context of gene regulation.Citation48 Thus, in RA derived T- and B-lymphocytes, hypermethylation was mostly observed at CpG island-associated sites, whereas hypomethylation occurred most frequently at CpGs within gene bodies. It is interesting to note that a similar predominance of differentially methylated CpG loci located within gene bodies was recently observed in joint derived cells from patients with RA.Citation28
A caveat associated with our findings is that they are based on a relatively small group of patients. However, all patients were female Caucasians with seropositive established disease, were all treated with methotrexate and were all non-smokers, which mitigates against the influence of potentially confounding variables, such as race, gender and smoking that have previously been associated with DNA methylation.Citation49-Citation52 Indeed, despite some variation in age, disease duration and the specific treatments received, the methylation changes at candidate CpGs did not appear to be related to these factors. Rather, candidate CpGs showed consistent methylation changes across multiple patient samples suggesting that the observed changes were indeed disease-related. To our knowledge, established relationships between the co-medications patients were receiving and effects on genome-wide DNA methylation have not been described. However, we are mindful that these medications and perhaps other unknown confounders may influence our findings. The findings from our work will require independent verification in larger patient cohorts, and perhaps to be extended to include specific lymphocyte subsets.
In summary, and by focusing on individual lymphocyte populations in this and our previous study,Citation22 we have identified intrinsic methylation differences between T- and B-lymphocytes that are consistent in both RA patients and healthy individuals. Moreover, using a genome-wide approach we report in patients with RA evidence of disease-associated aberrations of DNA methylation at multiple CpG loci that are distinct to either the T- or B-lymphocyte populations. These findings will form the basis for further investigations by us and other research groups to further explore the role of DNA methylation and other putative epigenetic changes in the genesis, progression and clinical management of RA.
Patients and Methods
Patients
Peripheral blood samples were collected from 12 Caucasian female patients with seropositive established RA (disease duration ≥ 5 y) and from 12 healthy Caucasian females who served as a control group. All RA patients satisfied the 1987 American College of Rheumatology revised criteria set for the classification of RACitation53 and were being treated with methotrexate as monotherapy (n = 5) or in combination with sulphasalazine (n = 6) or hydroxychloroquine (n = 1). Six patients were receiving non-steroidal anti-inflammatory drugs and three were taking analgesics. A number of patients were also receiving medication for co-morbid diseases, including hypertension (n = 4), hypercholesterolemia (n = 2), type II diabetes (n = 2), anemia (n = 1) and osteoporosis (n = 1). Five patients had no documented co-morbidities and no patients had been diagnosed with cancer at the time of recruitment. The average age and disease duration of patients was 64.6 ± 7.5 y and 15.8 ± 10.7 y (mean ± SD), respectively. Health status for the control group was established using a short questionnaire, excluding any individuals reporting inflammatory or musculoskeletal-related conditions. The average age for the healthy control group was 52.6 ± 4.9 y (mean ± SD). This was significantly lower than the average age of the RA group (P < 0.01, Student’s t test). All subjects were non-smokers. Subjects were recruited at the Haywood Rheumatology Centre in Stoke-on-Trent, UK and written informed consent was obtained in all cases. The study was approved by the East Midlands (Derby) Research Ethics Committee.
Purification of T- and B-lymphocyte populations
Peripheral blood mononuclear cells were isolated from whole blood using density-gradient centrifugation (Histopaque-1077, Sigma-Aldrich), following which CD3+ T-lymphocytes and CD19+ B-lymphocyte populations were purified by positive selection using anti-CD3 and anti-CD19 magnetic microbeads, respectively, and according to the manufacturer’s instructions (MiniMACS® Separation System; Miltenyi Biotec). Isolated cell populations were lysed, homogenized (QIAShredder spin columns; Qiagen) and archived prior to DNA extraction. An aliquot of each purified T- and B-lymphocyte population was stained with either an anti-CD3 or anti-CD20 phycoerythrin-labeled IgG monoclonal antibody (Miltenyi Biotec), respectively, for assessment of cell purity by flow cytometry, as we have previously described.Citation22 T-lymphocyte and B-lymphocyte purities were 99.1 ± 0.1% and 89.5 ± 11.3% respectively for RA patients, and 99.5 ± 0.4% and 90.9 ± 9.5% respectively for the healthy control group (mean ± SD).
Genome-wide DNA methylation profiling
Genome-wide DNA methylation profiling was performed using Infinium-based HumanMethylation450 BeadChips (Illumina Inc.) that interrogate over 480,000 unique CpG sites across the genome (99% of RefSeq genes).Citation54 Genomic DNA was first extracted from cell lysates using an AllPrep DNA/RNA Mini kit (Qiagen). Subsequently, T- and B-lymphocyte DNA from the 12 RA patients and 12 healthy subjects (48 samples in total) were sodium bisulfite converted and hybridized to arrays according to Illumina recommended protocols (performed by Gen-Probe Life Sciences Ltd) that we have previously described.Citation22 Methylation at individual CpGs is reported as a methylation β-value,Citation54 which falls on a continuous linear scale ranging from 0 (unmethylated) to 1 (completely methylated).
Validation by sodium bisulfite pyrosequencing
Validation of array data and independent quantification of methylation at candidate CpG loci was performed by sodium bisulfite pyrosequencing. To increase template quantity for pyrosequencing assays, we first performed whole genome amplification of bisulfite-converted DNA according to conditions we have recently described.Citation22 Thereafter, amplicons containing CpGs of interest were prepared by a touchdown PCR methodCitation55 using forward, reverse and sequencing primers specific for bisulfite-converted DNA (designed using PyroMark Assay Design software (Qiagen) and purchased from Biomers.net) (assay details are provided in Table S2). Pyrosequencing assays were run on a PyroMark Q24 instrument according to the manufacturer’s instructions (Qiagen), and in each case included at least one control dispensation to confirm sequence identity and completeness of bisulfite conversion. Pyrograms were analyzed using PyroMark Q24 software (v 2.0.6., build 20; Qiagen).
Data analysis
Initial processing, probe type correction and assessment of array data was conducted using NIMBL software.Citation56 To adjust for the reported difference in sensitivity between the two probe types on the arrays, NIMBL was used to perform ‘peak-based correction’ according to Dedeurwaerder et al.Citation57 All comparative analyses were conducted on peak-based corrected β-values. The Illumina 450k array allows parallel assessment of 12 samples reducing within-array variability. However, the effect of batch (array-to-array) variation on Illumina 450k arrays is still debated and a consensus on an appropriate method to “normalize” data has not been reached.Citation58-Citation60 In an attempt to best control for batch-effects, array experiments were conducted with block-randomization such that six RA and six healthy samples were present on each of the arrays. Each array passed quality control assessment based on the performance of internal array controls, and the distribution of β-values across all array CpGs was found to be similar in each of the T- and B-lymphocyte samples. Subsequent to array hybridization, it was necessary to exclude one healthy individual who no longer satisfied the clinical exclusion criteria. All analyses were thus conducted with a revised group of 11 healthy subjects. For each of the T- and B-lymphocyte data sets separately, and as we have previously described,Citation22 we excluded all CpGs for which one or more of the 23 samples displayed detection p-values > 0.05 (indicating an unreliable site) or presented with missing β-values. In performing this step, we excluded 1.7% and 1.1% of array CpGs from the T-lymphocyte and B-lymphocyte data sets and therefore retained 477,312 and 479,639 CpGs, respectively. Criteria for identification of CpGs displaying altered methylation between RA patients and healthy subjects for each cell type are described in the Results. Hierarchical clustering using Euclidian distance and average linkage criteria was performed using Genesis software (v1.7.6)Citation61 and statistical analyses were performed using NCSS 2000 (NCSS LCC). P values < 0.05 were considered significant.
| Abbreviations: | ||
| RA | = | rheumatoid arthritis |
| CpG | = | cytosine-guanine dinucleotide |
| Th | = | T-helper |
| IL | = | interleukin |
| 450K array/array | = | HumanMethylation450 BeadChip |
| NIMBL | = | Numerical Identification of Methylation Biomarker Lists |
| DAVID | = | database for annotation, visualization and integrated discovery |
| LINE-1 | = | long interspersed element-1 |
| MGMT | = | O-6-methylguanine-DNA methyltransferase |
| CCS | = | copper chaperone for superoxide dismutase |
| DUSP22 | = | dual specificity phosphatase 22 |
| GALNT9 | = | UDP-N-acetyl-alpha-D-galactosamine:polypeptide N-acetylgalactosaminyltransferase 9 |
Additional material
Download Zip (813 KB)Conflict-of-interest and financial disclosure statement
All listed authors (JRG, RDE, NBN, KEH, JCP, PTD, AAF, DLM and WEF) have no conflicts-of-interest to declare, and have no relevant financial relationships to disclose.
Acknowledgments
This work was supported by funding provided by the Haywood Rheumatism Research and Development Foundation.
References
- Cedar H, Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet 2009; 10:295 - 304; http://dx.doi.org/10.1038/nrg2540; PMID: 19308066
- Javierre BM, Hernando H, Ballestar E. Environmental triggers and epigenetic deregulation in autoimmune disease. Discov Med 2011; 12:535 - 45; PMID: 22204770
- Bottini N, Firestein GS. Epigenetics in rheumatoid arthritis: a primer for rheumatologists. Curr Rheumatol Rep 2013; 15:372; http://dx.doi.org/10.1007/s11926-013-0372-9; PMID: 24072602
- Ballestar E. Epigenetic alterations in autoimmune rheumatic diseases. Nat Rev Rheumatol 2011; 7:263 - 71; http://dx.doi.org/10.1038/nrrheum.2011.16; PMID: 21343899
- Wilson CB, Rowell E, Sekimata M. Epigenetic control of T-helper-cell differentiation. Nat Rev Immunol 2009; 9:91 - 105; http://dx.doi.org/10.1038/nri2487; PMID: 19151746
- Garaud S, Youinou P, Renaudineau Y. DNA methylation and B-cell autoreactivity. Adv Exp Med Biol 2011; 711:50 - 60; http://dx.doi.org/10.1007/978-1-4419-8216-2_5; PMID: 21627042
- Bird A. DNA methylation patterns and epigenetic memory. Genes Dev 2002; 16:6 - 21; http://dx.doi.org/10.1101/gad.947102; PMID: 11782440
- Ansel KM, Lee DU, Rao A. An epigenetic view of helper T cell differentiation. Nat Immunol 2003; 4:616 - 23; http://dx.doi.org/10.1038/ni0703-616; PMID: 12830136
- Agarwal S, Rao A. Modulation of chromatin structure regulates cytokine gene expression during T cell differentiation. Immunity 1998; 9:765 - 75; http://dx.doi.org/10.1016/S1074-7613(00)80642-1; PMID: 9881967
- Shin HJ, Park HY, Jeong SJ, Park HW, Kim YK, Cho SH, Kim YY, Cho ML, Kim HY, Min KU, et al. STAT4 expression in human T cells is regulated by DNA methylation but not by promoter polymorphism. J Immunol 2005; 175:7143 - 50; http://dx.doi.org/10.4049/jimmunol.175.11.7143; PMID: 16301617
- Kim EG, Shin HJ, Lee CG, Park HY, Kim YK, Park HW, Cho SH, Min KU, Cho ML, Park SH, et al. DNA methylation and not allelic variation regulates STAT6 expression in human T cells. Clin Exp Med 2010; 10:143 - 52; http://dx.doi.org/10.1007/s10238-009-0083-8; PMID: 19949830
- Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol 2008; 8:523 - 32; http://dx.doi.org/10.1038/nri2343; PMID: 18566595
- Polansky JK, Kretschmer K, Freyer J, Floess S, Garbe A, Baron U, Olek S, Hamann A, von Boehmer H, Huehn J. DNA methylation controls Foxp3 gene expression. Eur J Immunol 2008; 38:1654 - 63; http://dx.doi.org/10.1002/eji.200838105; PMID: 18493985
- Richardson B, Scheinbart L, Strahler J, Gross L, Hanash S, Johnson M. Evidence for impaired T cell DNA methylation in systemic lupus erythematosus and rheumatoid arthritis. Arthritis Rheum 1990; 33:1665 - 73; http://dx.doi.org/10.1002/art.1780331109; PMID: 2242063
- Liu CC, Fang TJ, Ou TT, Wu CC, Li RN, Lin YC, Lin CH, Tsai WC, Liu HW, Yen JH. Global DNA methylation, DNMT1, and MBD2 in patients with rheumatoid arthritis. Immunol Lett 2011; 135:96 - 9; http://dx.doi.org/10.1016/j.imlet.2010.10.003; PMID: 20937307
- Nile CJ, Read RC, Akil M, Duff GW, Wilson AG. Methylation status of a single CpG site in the IL6 promoter is related to IL6 messenger RNA levels and rheumatoid arthritis. Arthritis Rheum 2008; 58:2686 - 93; http://dx.doi.org/10.1002/art.23758; PMID: 18759290
- Fu LH, Ma CL, Cong B, Li SJ, Chen HY, Zhang JG. Hypomethylation of proximal CpG motif of interleukin-10 promoter regulates its expression in human rheumatoid arthritis. Acta Pharmacol Sin 2011; 32:1373 - 80; http://dx.doi.org/10.1038/aps.2011.98; PMID: 21986577
- Kitamura T, Kabuyama Y, Kamataki A, Homma MK, Kobayashi H, Aota S, Kikuchi S, Homma Y. Enhancement of lymphocyte migration and cytokine production by ephrinB1 system in rheumatoid arthritis. Am J Physiol Cell Physiol 2008; 294:C189 - 96; http://dx.doi.org/10.1152/ajpcell.00314.2007; PMID: 17942634
- Liao J, Liang G, Xie S, Zhao H, Zuo X, Li F, Chen J, Zhao M, Chan TM, Lu Q. CD40L demethylation in CD4(+) T cells from women with rheumatoid arthritis. Clin Immunol 2012; 145:13 - 8; http://dx.doi.org/10.1016/j.clim.2012.07.006; PMID: 22889643
- Jeffries MA, Dozmorov M, Tang Y, Merrill JT, Wren JD, Sawalha AH. Genome-wide DNA methylation patterns in CD4+ T cells from patients with systemic lupus erythematosus. Epigenetics 2011; 6:593 - 601; http://dx.doi.org/10.4161/epi.6.5.15374; PMID: 21436623
- Ellis JA, Munro JE, Chavez RA, Gordon L, Joo JE, Akikusa JD, Allen RC, Ponsonby AL, Craig JM, Saffery R. Genome-scale case-control analysis of CD4+ T-cell DNA methylation in juvenile idiopathic arthritis reveals potential targets involved in disease. Clin Epigenetics 2012; 4:20; http://dx.doi.org/10.1186/1868-7083-4-20; PMID: 23148518
- Glossop JR, Nixon NB, Emes RD, Haworth KE, Packham JC, Dawes PT, Fryer AA, Mattey DL, Farrell WE. Epigenome-wide profiling identifies significant differences in DNA methylation between matched-pairs of T- and B-lymphocytes from healthy individuals. Epigenetics 2013; 8:1188 - 97; http://dx.doi.org/10.4161/epi.26265; PMID: 24005183
- Roessler J, Ammerpohl O, Gutwein J, Hasemeier B, Anwar SL, Kreipe H, Lehmann U. Quantitative cross-validation and content analysis of the 450k DNA methylation array from Illumina, Inc. BMC Res Notes 2012; 5:210; http://dx.doi.org/10.1186/1756-0500-5-210; PMID: 22546179
- Byun HM, Siegmund KD, Pan F, Weisenberger DJ, Kanel G, Laird PW, Yang AS. Epigenetic profiling of somatic tissues from human autopsy specimens identifies tissue- and individual-specific DNA methylation patterns. Hum Mol Genet 2009; 18:4808 - 17; http://dx.doi.org/10.1093/hmg/ddp445; PMID: 19776032
- Duong CV, Emes RD, Wessely F, Yacqub-Usman K, Clayton RN, Farrell WE. Quantitative, genome-wide analysis of the DNA methylome in sporadic pituitary adenomas. Endocr Relat Cancer 2012; 19:805 - 16; http://dx.doi.org/10.1530/ERC-12-0251; PMID: 23045325
- Takami N, Osawa K, Miura Y, Komai K, Taniguchi M, Shiraishi M, Sato K, Iguchi T, Shiozawa K, Hashiramoto A, et al. Hypermethylated promoter region of DR3, the death receptor 3 gene, in rheumatoid arthritis synovial cells. Arthritis Rheum 2006; 54:779 - 87; http://dx.doi.org/10.1002/art.21637; PMID: 16508942
- Karouzakis E, Rengel Y, Jüngel A, Kolling C, Gay RE, Michel BA, Tak PP, Gay S, Neidhart M, Ospelt C. DNA methylation regulates the expression of CXCL12 in rheumatoid arthritis synovial fibroblasts. Genes Immun 2011; 12:643 - 52; http://dx.doi.org/10.1038/gene.2011.45; PMID: 21753787
- Nakano K, Whitaker JW, Boyle DL, Wang W, Firestein GS. DNA methylome signature in rheumatoid arthritis. Ann Rheum Dis 2013; 72:110 - 7; http://dx.doi.org/10.1136/annrheumdis-2012-201526; PMID: 22736089
- de la Rica L, Urquiza JM, Gómez-Cabrero D, Islam AB, López-Bigas N, Tegnér J, Toes RE, Ballestar E. Identification of novel markers in rheumatoid arthritis through integrated analysis of DNA methylation and microRNA expression. J Autoimmun 2013; 41:6 - 16; http://dx.doi.org/10.1016/j.jaut.2012.12.005; PMID: 23306098
- Liu Y, Aryee MJ, Padyukov L, Fallin MD, Hesselberg E, Runarsson A, Reinius L, Acevedo N, Taub M, Ronninger M, et al. Epigenome-wide association data implicate DNA methylation as an intermediary of genetic risk in rheumatoid arthritis. Nat Biotechnol 2013; 31:142 - 7; http://dx.doi.org/10.1038/nbt.2487; PMID: 23334450
- Reinius LE, Acevedo N, Joerink M, Pershagen G, Dahlén SE, Greco D, Söderhäll C, Scheynius A, Kere J. Differential DNA methylation in purified human blood cells: implications for cell lineage and studies on disease susceptibility. PLoS One 2012; 7:e41361; http://dx.doi.org/10.1371/journal.pone.0041361; PMID: 22848472
- Cope AP. T cells in rheumatoid arthritis. Arthritis Res Ther 2008; 10:Suppl 1 S1; http://dx.doi.org/10.1186/ar2412; PMID: 19007421
- Martinez-Gamboa L, Brezinschek HP, Burmester GR, Dörner T. Immunopathologic role of B lymphocytes in rheumatoid arthritis: rationale of B cell-directed therapy. Autoimmun Rev 2006; 5:437 - 42; http://dx.doi.org/10.1016/j.autrev.2006.02.004; PMID: 16920569
- Sekine Y, Tsuji S, Ikeda O, Sato N, Aoki N, Aoyama K, Sugiyama K, Matsuda T. Regulation of STAT3-mediated signaling by LMW-DSP2. Oncogene 2006; 25:5801 - 6; http://dx.doi.org/10.1038/sj.onc.1209578; PMID: 16636663
- Fonseca JE, Santos MJ, Canhão H, Choy E. Interleukin-6 as a key player in systemic inflammation and joint destruction. Autoimmun Rev 2009; 8:538 - 42; http://dx.doi.org/10.1016/j.autrev.2009.01.012; PMID: 19189867
- Shouda T, Yoshida T, Hanada T, Wakioka T, Oishi M, Miyoshi K, Komiya S, Kosai K, Hanakawa Y, Hashimoto K, et al. Induction of the cytokine signal regulator SOCS3/CIS3 as a therapeutic strategy for treating inflammatory arthritis. J Clin Invest 2001; 108:1781 - 8; http://dx.doi.org/10.1172/JCI13568; PMID: 11748261
- Mori T, Miyamoto T, Yoshida H, Asakawa M, Kawasumi M, Kobayashi T, Morioka H, Chiba K, Toyama Y, Yoshimura A. IL-1β and TNFα-initiated IL-6-STAT3 pathway is critical in mediating inflammatory cytokines and RANKL expression in inflammatory arthritis. Int Immunol 2011; 23:701 - 12; http://dx.doi.org/10.1093/intimm/dxr077; PMID: 21937456
- Anderson A, Routledge C, Isaacs J, Pratt A. Investigation of interleukin-6-driven STAT3 signalling in circulating lymphocytes of patients with early rheumatoid arthritis as a route to biomarker discovery. Lancet 2014; 383:S84; http://dx.doi.org/10.1016/S0140-6736(14)60347-0
- Pratt AG, Swan DC, Richardson S, Wilson G, Hilkens CM, Young DA, Isaacs JD. A CD4 T cell gene signature for early rheumatoid arthritis implicates interleukin 6-mediated STAT3 signalling, particularly in anti-citrullinated peptide antibody-negative disease. Ann Rheum Dis 2012; 71:1374 - 81; http://dx.doi.org/10.1136/annrheumdis-2011-200968; PMID: 22532634
- Altorok N, Coit P, Hughes T, Koelsch KA, Stone DU, Rasmussen A, Radfar L, Scofield RH, Sivils KL, Farris AD, et al. Genome-wide DNA methylation patterns in naive CD4+ T cells from patients with primary Sjögren’s syndrome. Arthritis Rheumatol 2014; 66:731 - 9; http://dx.doi.org/10.1002/art.38264; PMID: 24574234
- Ten Hagen KG, Fritz TA, Tabak LA. All in the family: the UDP-GalNAc:polypeptide N-acetylgalactosaminyltransferases. Glycobiology 2003; 13:1R - 16R; http://dx.doi.org/10.1093/glycob/cwg007; PMID: 12634319
- Ishino H, Kawahito Y, Hamaguchi M, Takeuchi N, Tokunaga D, Hojo T, Wada M, Yamamoto A, Kadoya M, Tsubouchi Y, et al. Expression of Tn and sialyl Tn antigens in synovial tissues in rheumatoid arthritis. Clin Exp Rheumatol 2010; 28:246 - 9; PMID: 20483047
- Hamaguchi M, Kawahito Y, Ishino H, Takeuchi N, Tokunaga D, Hojo T, Yamamoto A, Kadoya M, Seno T, Kohno M, et al. Mucin from rheumatoid arthritis synovial fluid enhances interleukin-6 production by human peripheral blood mononuclear cells. Hum Immunol 2011; 72:241 - 8; http://dx.doi.org/10.1016/j.humimm.2010.12.013; PMID: 21195737
- Rauch TA, Wu X, Zhong X, Riggs AD, Pfeifer GP. A human B cell methylome at 100-base pair resolution. Proc Natl Acad Sci U S A 2009; 106:671 - 8; http://dx.doi.org/10.1073/pnas.0812399106; PMID: 19139413
- Jjingo D, Conley AB, Yi SV, Lunyak VV, Jordan IK. On the presence and role of human gene-body DNA methylation. Oncotarget 2012; 3:462 - 74; PMID: 22577155
- Bird AP. CpG-rich islands and the function of DNA methylation. Nature 1986; 321:209 - 13; http://dx.doi.org/10.1038/321209a0; PMID: 2423876
- Antequera F, Bird A. Number of CpG islands and genes in human and mouse. Proc Natl Acad Sci U S A 1993; 90:11995 - 9; http://dx.doi.org/10.1073/pnas.90.24.11995; PMID: 7505451
- Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, Cui H, Gabo K, Rongione M, Webster M, et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet 2009; 41:178 - 86; http://dx.doi.org/10.1038/ng.298; PMID: 19151715
- Liu J, Morgan M, Hutchison K, Calhoun VD. A study of the influence of sex on genome wide methylation. PLoS One 2010; 5:e10028; http://dx.doi.org/10.1371/journal.pone.0010028; PMID: 20386599
- Zhang FF, Cardarelli R, Carroll J, Fulda KG, Kaur M, Gonzalez K, Vishwanatha JK, Santella RM, Morabia A. Significant differences in global genomic DNA methylation by gender and race/ethnicity in peripheral blood. Epigenetics 2011; 6:623 - 9; http://dx.doi.org/10.4161/epi.6.5.15335; PMID: 21739720
- Breitling LP, Yang R, Korn B, Burwinkel B, Brenner H. Tobacco-smoking-related differential DNA methylation: 27K discovery and replication. Am J Hum Genet 2011; 88:450 - 7; http://dx.doi.org/10.1016/j.ajhg.2011.03.003; PMID: 21457905
- Zöchbauer-Müller S, Lam S, Toyooka S, Virmani AK, Toyooka KO, Seidl S, Minna JD, Gazdar AF. Aberrant methylation of multiple genes in the upper aerodigestive tract epithelium of heavy smokers. Int J Cancer 2003; 107:612 - 6; http://dx.doi.org/10.1002/ijc.11458; PMID: 14520700
- Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, Healey LA, Kaplan SR, Liang MH, Luthra HS, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum 1988; 31:315 - 24; http://dx.doi.org/10.1002/art.1780310302; PMID: 3358796
- Bibikova M, Barnes B, Tsan C, Ho V, Klotzle B, Le JM, Delano D, Zhang L, Schroth GP, Gunderson KL, et al. High density DNA methylation array with single CpG site resolution. Genomics 2011; 98:288 - 95; http://dx.doi.org/10.1016/j.ygeno.2011.07.007; PMID: 21839163
- Haworth KE, Farrell WE, Emes RD, Ismail KM, Carroll WD, Borthwick HA, Yates AM, Hubball E, Rooney A, Khanam M, et al. Combined influence of gene-specific cord blood methylation and maternal smoking habit on birth weight. Epigenomics 2013; 5:37 - 49; http://dx.doi.org/10.2217/epi.12.72; PMID: 23414319
- Wessely F, Emes RD. Identification of DNA methylation biomarkers from Infinium arrays. Front Genet 2012; 3:161; http://dx.doi.org/10.3389/fgene.2012.00161; PMID: 22936948
- Dedeurwaerder S, Defrance M, Calonne E, Denis H, Sotiriou C, Fuks F. Evaluation of the Infinium Methylation 450K technology. Epigenomics 2011; 3:771 - 84; http://dx.doi.org/10.2217/epi.11.105; PMID: 22126295
- Pidsley R, Y Wong CC, Volta M, Lunnon K, Mill J, Schalkwyk LC. A data-driven approach to preprocessing Illumina 450K methylation array data. BMC Genomics 2013; 14:293; http://dx.doi.org/10.1186/1471-2164-14-293; PMID: 23631413
- Touleimat N, Tost J. Complete pipeline for Infinium(®) Human Methylation 450K BeadChip data processing using subset quantile normalization for accurate DNA methylation estimation. Epigenomics 2012; 4:325 - 41; http://dx.doi.org/10.2217/epi.12.21; PMID: 22690668
- Pan H, Chen L, Dogra S, Teh AL, Tan JH, Lim YI, Lim YC, Jin S, Lee YK, Ng PY, et al. Measuring the methylome in clinical samples: improved processing of the Infinium Human Methylation450 BeadChip Array. Epigenetics 2012; 7:1173 - 87; http://dx.doi.org/10.4161/epi.22102; PMID: 22964528
- Sturn A, Quackenbush J, Trajanoski Z. Genesis: cluster analysis of microarray data. Bioinformatics 2002; 18:207 - 8; http://dx.doi.org/10.1093/bioinformatics/18.1.207; PMID: 11836235