
Abstract
We performed a genome-wide analysis of aberrant DNA methylation in chronic lymphocytic leukemia (CLL) using methylated CpG island amplification (MCA) coupled with a promoter microarray. We identified 280 potential targets of aberrant DNA methylation in CLL. These genes were located more frequently in chromosomes 19 (16%, p=0.001), 16 (11%, p=0.001), 17 (10%, p=0.02) and 11 (9%, p=0.02) and could be grouped in several functional networks. Methylation status was confirmed for 22 of these genes (SOX11, DLX1, FAM62C, SOX14, RSPO1, ADCY5, HAND2,SPOCK, MLL, ING1, PRIMA1, BCL11B, LTBP2, BNC1, NR2F2, SALL1, GALGT2, LHX1, DLX4, KLK10, TFAP2 and APP) in 78 CLL patients by pyrosequencing. As a proof of principle, we analyzed the expression of 2 genes, PRIMA1 and APP, in primary cells and of GALGT2, TFAP2C and PRIMA1 in leukemia cells. There was an inverse association between methylation and gene expression. This could be reversed by treatment with 5-aza-2’-deoxycytidine in cell lines. Treatment in a clinical trial with 5-azacitidine resulted in decreased methylation of LINE, DLX4 and SALL1 in the peripheral blood B-cells of patients with CLL. IgVH mutational status or ZAP-70 expression were not associated with specific methylation profiles. By multivariate analysis, methylation of LINE and APP was associated with shorter overall survival (p = 0.045 and 0.0035, respectively). This study demonstrates that aberrant DNA methylation is common and has potential prognostic and therapeutic value in CLL.
Acknowledgements
This work was supported by the CLL Global Research Foundation. G.G.-M. is also funded by grants CA100067 and CA105771 from the NCI, a Physician-Scientist Award from the Commonwealth Foundation for Cancer Research at the University of Texas M.D. Anderson Cancer Center and the Leukemia and Lymphoma Society of America. This work was also supported by a grant from Chronic Lymphocytic Leukemia Foundation to W.-G.T.
Figures and Tables
Figure 1 Heatmap representations of methylation profiles of 22 candidate genes. Methylation was measured in patients with CLL (n = 78) and NBCs (n = 10) using bisulfite pyrosequencing. Green indicates a methylation density of <10%, yellow 10–49% and red ≥50%. White indicates lack of data due to failure of the assay for this particular sample. Tester includes the profile of the two patients with CLL and 17p deletion, driver includes the NBCs from two healthy volunteers whose DNA was used to perform the original MCA experiment. The methylation profiles of the two testers and two drivers are shown at the top of the figure.
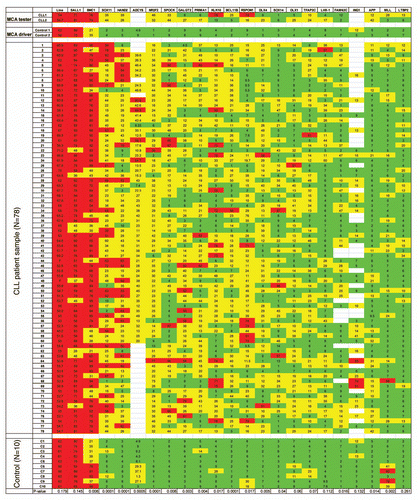
Figure 2 (A and B) Correlation between methylation (%) and gene expression (dCTt) of two selected genes, PRIMA1 and APP. Methylation was measured by bisulfite pyrosequencing and gene expression was measured by real-time RT-PCR assay. (C and D) Epigenetic modulation of GALGT2, TFAP2C and PRIMA1 gene expression in Raji and HL60 leukemia cell lines. Leukemia cells were treated with 5-aza-2′-deoxycytidine (DAC) at 1 µM for 4 days alone, or DAC at 1 µM for 4 days and then trichostatin A (TSA) at 500 nM for the last 24 h, or TSA at 500 nM for the last 24 h alone without pre-treatment with DAC. Medium was changed daily with fresh drugs, and gene expression was measured by real-time RT-PCR assay.
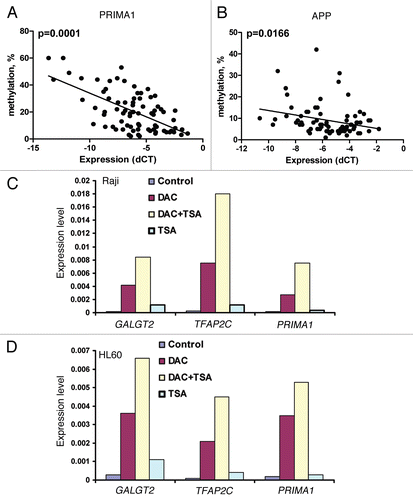
Figure 3 (A) In vivo effect of 5-azacitidine in patients with CLL. Changes in methylation of LINE, SOX11 and DLX4 genes were measured in two patients with CLL who were treated in a clinical trial with 5-azacitidine. DNA was isolated from peripheral blood mononuclear cells and the methylation was measured by bisulfite pyrosequencing at each time point indicated. C, cycle number; D, days of treatment. (B and C) Methylation of LINE, APP and overall survival. Kaplan-Meyer survival analysis showing both increased methylation of LINE (p = 0.045) and APP (p = 0.0035) were associated with shorter overall survival using optimal cut-points at 58.8 and 17%, respectively.
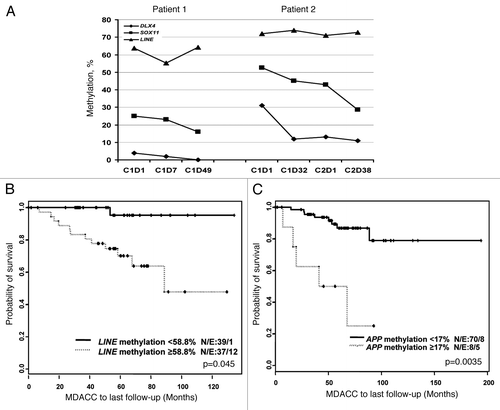
Figure 4 Hierarchic cluster analysis of methylation data and its correlation with IgVH mutational status (A) and ZAP-70 expression (B). Cluster analysis of methylation data was performed using Commoner application from National Cancer Institute (www.discover.nci.nih.gov/cimminer). Each column represents an individual patient and each row represents a gene.
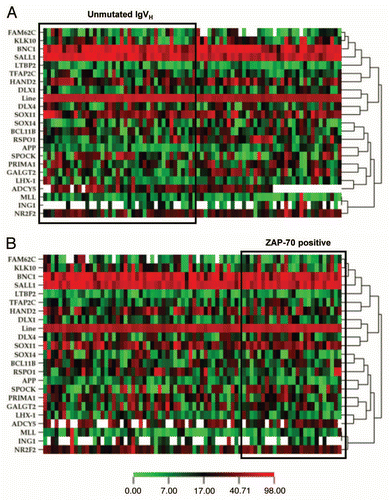
Table 1 Differences in gene methylation status by prognostic group
Table 2 Multivariable cox proportional hazard model for and survival