Abstract
The bromodomain is an evolutionarily conserved motif found in many transcriptional activators including p300 which contains an intrinsic histone acetyltransferase activity and is a general coactivator for many transcription factors. One mode of bromodomain action is to serve as a binding module to recognize specific acetyl-lysine residue of histones during chromatin remodeling and transcriptional activation. The function of p300 is required for diverse sets of gene expression. However, it is not known whether the p300 bromodomain is involved in the expression of all or only subset of p300-dependent genes. In this study, we examined the impact of either wild type or a bromo-deficient p300 on the expression of several p300-dependant genes. The effects of histone acetylation on the expression of these genes were also assessed by targeting histone deacetylase activities with an inhibitor approach. We show that the impact of these inhibitors on the transcriptional activation of p300-dependent genes are impaired in cells containing the bromo-deficient p300, indicating that the interplay of p300 and histone acetylation in p300-dependent gene transcription requires the bromodomain. We also observed an increase in the expression of bromo-deficient p300 at the level of transcription possibly to compensate for the loss of p300 function. However, the high level of bromo-deficient p300 is not able to maintain the basal level of histone acetylation. Thus, the bromodomain is important for p300 to maintain the basal level of histone acetylation and to induce the transcriptional activation of p300-dependent genes. Nevertheless, the requirement of bromodomain and histone acetylation in p300-dependent gene transcription is determined by a gene specific manner.
Introduction
Transcriptional coactivator p300 was identified initially as an E1A-associated protein and later as a tumor suppressor.Citation1,Citation2 Mutations in p300 genes have been detected in various epithelial cancers and reintroduction of wild-type p300 leads to growth suppression of the cancer cells.Citation3–Citation5 Patients with Rubinstein-Taybi syndrome due to p300 heterozygosity have an increased predisposition to cancer.Citation6–Citation8 In addition, E1A, HPV E6 oncoprotein and SV40 large T antigen target p300, further underlining a critical role for p300 in events related to cellular transformation.Citation9–Citation11
The p300 contains an intrinsic histone acetyltransferase (HAT) activity and multiple interaction surfaces for a variety of transcription factors and components of the basal transcription machinery.Citation12 It thus acts as either a HAT enzyme to acetylate histones or a bridging factor to integrate different regulatory signals in transcriptional activation.Citation5,Citation12 One of the p300 domains is an evolutionarily conserved bromodomain which was characterized first for the Drosophila melanogaster Brahma protein and later found in many transcription regulators and nearly all nuclear HAT proteins.Citation13,Citation14 The function of bromodomain is important for p300 to recognize specific chromatin substrates and to coordinate chromatin remodeling and transcriptional activation.Citation15 One mode of bromodomain action is to serve as acetyl-lysine binding module tethering the HAT activity to a defined chromosomal site to achieve highly specific histone acetylation.Citation16–Citation19 However, it is not clear whether the bromodomain is required for gene expression of all or only a limited set of p300-dependent genes.
The overall topology of the bromodomain modules is similar among P/CAF, GCN5 and TAFII250 and the residues important for acetyl-lysine recognition are largely conserved.Citation17,Citation20,Citation21 However, there is a clear difference in the ligand specificity of bromodomain. It is the bromodomain of P/CAF, not CBP, that binds specifically to the acetyl-lysine50 of HIV-1 Tat.Citation19 Likewise, the binding of CBP to acetyl-lysine382 of p53 is required for p53 acetylation-dependent recruitment of coactivator in response to UV-induced DNA damage, but the association of p300 with p53 does not depend on the bromodomains-acetyl-lysine interaction.Citation18 The bromodomain of p300 and CBP are highly homologous, yet they differ in ligand selectivity, indicating that the ligand selectivity is controlled by few but very important variations in bromodomain sequences or the action mode of the bromodomain is not solely linked to the acetyl-lysine binding.
Targeting histone deacetylase (HDAC) with inhibitors affects gene expression through inhibiting histone deacetylation, which results in the accumulation of acetylated histones and the association of activator complex to activate gene transcription.Citation22–Citation24 Therefore, HDAC inhibitors have emerged in recent years as a new class of cancer therapeutics since they selectively induce apoptosis in tumor cells, with limited toxicity to normal cells.Citation25 There are several classes of natural and synthetic HDAC inhibitors. One is the short-chain fatty acids such as butyrate and valproic acid.Citation26,Citation27 Butyrate is naturally produced by anaerobic bacteria fermenting undigested dietary carbohydrates and induces cell cycle arrest and selective apoptosis of tumor cells through altering histone acetylation.Citation23,Citation28 Valproic acid is commonly used in the treatment of epilepsy, bipolar disorder and migraines and also selectively induces transformed cells to undergo growth arrest and apoptosis.Citation29 Trichostatin A (TSA), a hydroxamate compound represents another class of HDAC inhibitors.Citation30
In this study, we examined the roles of bromodomain and histone acetylation in the expression of several p300-dependant genes such as p21, Egr1 and E2F1, to determine the interplay of bromodomain and histone acetylation in the regulation of p300-dependent genes.
Results
Bromo-deficient p300 of SiHa cells.
The SiHa cervical carcinoma cells contain a homozygous internal deletion of exons 15–18 in the p300 gene (p300Δex15–18) which gives rise to a p300 protein lacking the bromodomain (bromo-deficient).Citation5 Most importantly, the HAT activity of the bromodomain deficient p300 is considerably impaired, only about 30% remaining compared to the wildtype p300.Citation5 To probe for cellular mechanisms involved in the regulation of p300 function, we first characterized in details the bromo-deficient p300 in SiHa cells in parallel with the wild-type p300 in HeLa cervical carcinoma cells.
Western analysis demonstrated that the level of the truncated form of p300 in the SiHa cells was significantly higher than the wild-type p300 in the HeLa cells, about 5-fold ( and B). Quantitative real-time RT-PCR analysis revealed that indeed the mRNA level of bromo-deficient p300 in the SiHa cells was significantly higher than the wild-type p300 in the HeLa cells, about 10-fold (). In addition, a pulse-labeling protocol demonstrated that the rate of protein synthesis of the bromo-deficient p300 in the SiHa cells was significantly higher than the wild-type p300 in the HeLa cells, about 10 fold ().
To examine the impact of bromodomain on p300 turnover, we employed a pulse chase protocol to compare the metabolic stability of wild-type and bromo-deficient p300. Previous studies show that the half life of wild-type p300 is about 11–14 h in several cell lines.Citation31–Citation33 We determined that in the HeLa cells, the apparent half life of wild-type p300 is about 11 h as assessed by a pulse-chase protocol (). In the SiHa cells, the apparent half-life of the bromo-deficient p300 was about 10 h, which is in the similar range of the wild-type p300 in the HeLa cells (comparing with E). In addition, immunofluorescence microscopy showed that the bromo-deficient p300, as the wild-type p300, localized predominantly to the nucleus (). Taken together, these data suggest that the increase in the level of the truncated form of p300 protein in the SiHa cells may stem from a feed-back regulation to compensate for the loss of p300 HAT activity, through the augmentation of p300Δex15–18 gene expression, but not a decrease of the bromo-deficient p300 turnover.
HDAC inhibitors and p300-dependent gene expression.
Genome-wide survey of promoter structure occupied by p300 has identified a group of p300-dependent genes, such as p21, Egr1 and E2F1, which contain the characteristic of GC rich region and some common motifs for transcription factor binding.Citation34 However, gene transcription of p21 and E2F1 in HeLa cells harboring a wild-type p300 exhibits differential sensitivity to treatment with HDAC inhibitor butyrate.Citation35
To define the roles of p300 and histone acetylation in the regulation of p300-dependent genes, we first examined the expression profile of p21, Egr1 and E2F1 following targeting HDAC activities with HDAC inhibitors such as butyrate, valproic acid and TSA in time course studies. As shown in and B, following 8 h of treatments with these HDAC inhibitors, the protein levels of p21 in the HeLa cells were significantly augmented, up to 7-fold, whereas 16 h of treatments increased the levels of p21 protein further to about 17-fold. Similarly, the protein levels of Egr1 were also significantly augmented, up to 3-fold following 8 h of treatment, and up to 9-fold following 16 h of treatments ( and B). On the other hand, the protein levels of E2F1 were not much affected by these treatments ( and B), which is consistent with previous report.Citation35 Taken together, our data suggest that the role of histone acetylation in the expression of p300-dependent genes may be determined by a gene specific manner.
Gene expression in cells harboring the bromo-deficient p300.
To examine the interplay of bromodomain and histone acetylation in the regulation of p300-depedent genes, we next examined the effects of butyrate, valproic acid and TSA, on the protein levels of p21, Egr1 and E2F1 in the SiHa cells containing the bromodeficient p300. Following 16 h of treatments, an increase in the levels of p21 protein was detected by western blotting analysis ( and D). However, this augmentation was only about 4-fold, significantly lower than the 17-fold increase seen in the HeLa cells (comparing with B). Similarly, the protein levels of Egr1 were only increased about 2-fold in response to the treatments ( and D), in contrast to the 9-fold increase seen in the HeLa cells (comparing with B). Interestingly, the protein levels of E2F1 were not affected by the treatments, which is the same as seen in the HeLa cells (comparing with B). Taken together, these data suggest that the interplay of p300 function and histone acetylation in the expression of p300-dependent genes may require the bromodomain.
Effects of HDAC inhibitors on the transcription of p300-dependent genes.
To determine if the effects of these HDAC inhibitors on the protein levels of p21 and Egr1 are transcription dependent, we profiled the mRNA levels of p21 and Egr1 following HDAC inhibitor treatments by using quantitative real time RT-PCR analysis. As shown in , treatments of the HeLa cells with butyrate, valproic acid or TSA for 4 h resulted in a significant increase in the levels of p21 mRNA, up to 3-fold, while 16 h of treatment further increased the augmentation to about 6-fold. Likewise, the levels of Egr1 mRNA were augmented significantly to about 4-fold following 4 h of treatments, and increased further to about 21-fold following 16 h of treatments (). Together, these results demonstrate that HDAC inhibitors increase the expression of p300-dependent genes such as p21 and Egr1 at the level of transcription.
Next we examined the transcript levels of p21 and Egr1 in the SiHa cells in response to these treatments. As shown in , following 16 h of treatments, the mRNA levels of p21 were only increased up to 3-fold, significantly lower than the 6-fold of increase observed in the HeLa cells (compare with A). With respect to Egr1, 16 h of treatments resulted in about 6-fold of augmentation in the levels of Egr1 mRNA, again significantly lower than the 21-fold of increase observed in the HeLa cells ( and B). Taken together, these data indicate that the impact of these HDAC inhibitors on transcription of p300-dependent genes is impaired in the SiHa cells which harbor a bromo-deficient p300.
Global histone acetylation and p300-dependent gene expression.
To determine the status of histone acetylation, we examined the effects of HDAC inhibitors on the levels of global histone acetylation in the HeLa and SiHa cells. The cells were treated with butyrate, valproic acid or TSA for 4 h and then subjected to western analysis with a specific antibody against acetylated K9 and K14 on histone H3. As shown in and B, treatments with these HDAC inhibitors increased the levels of acetylated H3 similarly, about 2-fold, in both the HeLa and SiHa cells ( and B). However, the basal level of H3 acetylation was significantly lower in the SiHa than the HeLa cells (). These data suggest that the bromodomain is important for p300 to exert its HAT activity in histone acetylation since the high level of bromo-deficient p300 was not able to maintain the basal level of histone acetylation in the SiHa cells ( and ).
We next examined the interplay bromodomain and histone acetylation in p300-depedent gene expression of SiHa cells. The cells were first transfected with expression plasmid for the wild-type p300 and then treated with butyrate, valproic acid or TSA for 8 h. As shown with the western analysis, p21 levels were upregulated by butyrate and TSA, whereas Egr1 levels were augmented by valproic acid and TSA (). Interestingly, the sensitivity of p21 or Egr1 to these HDAC inhibitors became similar to the HeLa cells in which butyrate is a strong inducer for p21 and valproic acid for Egr1 (comparing to ). Thus, these data suggest that the bromodomain is important for p300 to induce the transcriptional activation of p300-dependent genes.
Discussion
The major conclusion of this work is that the bromodomain is important for p300 to exert its HAT activity to maintain the basal level of histone acetylation and to induce the transcriptional activation of p300-dependent genes. However, the roles of bromodomain and histone acetylation in p300-dependent gene expression are determined in a gene specific manner.
Histone acetylation is generally associated with active gene loci and the acetylation of histone by p300 is an important avenue to achieve transcriptional activation. The acetylation of specific lysine residues on the histone tails plays distinct roles in gene regulation by marking the histone as a flag for transactivation.Citation36 In this event, p300 is often recruited to the regulatory elements by chromatin-remodeling factors and gene specific transcription factors. On the other hand, histone acetylation also serves as a binding signal for transcriptional coactivators such as p300.Citation17 In such case, the acetylated lysine residues are recognized and bound by the bromodomain to initiate the assembly of transcription factors and RNA polymerase II complex.
Here, we demonstrate that although histone acetylation is generally associated with transcriptional activation, it only regulates a subset of p300-dependent genes. HDAC inhibitors upregulate the expression of p21 and Egr1 in a time dependant manner in the presence of wild-type p300 ( and ), consistent with previous studies.Citation37,Citation38 However, the expression of E2F1 is not affected by HDAC inhibitors (), suggesting that p300 regulates E2F1 gene expression through a mechanism that is independent of histone acetylation. Interestingly, the augmentation of p21 and Egr1 expression by HDAC inhibitors is impaired in cells harboring a bromo-deficient p300 ( and ), indicating that the bromodomain function is important for transcriptional activation at the p21 and Egr1 promoter. The bromo-deficient p300 of SiHa cells was first identified by Ohshima et al.Citation39 They also show that the constitutively active TGFβ fails to induce the activation of p21 reporter in the SiHa cells. However, TGFβ is able to induce the transactivation of the p21 reporter upon reintroduction of the wild-type p300 into the cells, demonstrating that the bromodomain is important for p300 to recognize the acetylated histones as a coactivator at the p21 promoter.
The bromodomain is commonly found in transcriptional regulators, particularly in the nuclear HAT enzymes. Understandably, its function has been linked to acetyl-lysine recognition and chromatin remodeling.Citation16 The bromodomain of p300 is important for its full coactivator activity and mediates to a certain extent the stable association of p300 with chromatin template, which is essential for p300 function.Citation15,Citation40 The deletion of bromodomain reduces significantly the ability of p300 to interact with chromatin, since the bromodomain recognizes and binds to acetylated lysine 9 and 14 on the histone H3 tails.Citation40,Citation41 The acetylation levels of these lysines are augmented by HDAC inhibitors regardless of p300 functionality (). However, the expression of p21 and Egr1 gene is robustly induced by the HDAC inhibitor in cells containing the wild-type p300, but not the bromodomain-deficient p300 ( and ). Therefore, the bromodomain is required for the interplay of p300 and histone acetylation in the transcriptional activation of these p300-dependent genes.
The bromodomain is also required for maintaining p300 HAT activity. The loss of p300 bromodomain in the SiHa cells severely impairs the HAT activity.Citation5 Interestingly the basal level of histone acetylation in cells containing the bromo-deficient p300 is significantly lower than in cells containing the wild-type p300 cells (). One contribution of bromodomain to histone acetylation is by tethering the HATs to specific sites at the chromatin and stabilizing the HAT/chromatin remodeling complex.Citation36 It has been shown that bromo-deficient p300 is able to acetylate free histones but not the nucleosome histones in native chromatin.Citation42 Therefore, the bromodomain can act as an anchoring domain which tethers p300 to the chromatin template for histone acetylation, chromatin remodeling and transcriptional activation.Citation43
Feedback is an important regulatory mechanism to control the expression of genes or regulatory net works. We observed that the p300Δex15–18 gene transcription and translation in the SiHa cells are significantly higher than the wild-type p300 in the HeLa cells, which renders an abundance of bromo-deficient p300 protein in the SiHa cells (). This is most likely a result of feedback regulation to compensate for the loss of p300 HAT activity. Interestingly, the stability of bromo-deficient p300 is similar to the wild-type p300. Our data suggests a new mechanism for the bromodomain involved control of p300 function, through a functional feedback regulation of p300 gene expression, but not a p300 turnover.
The conclusion of this study is that the bromodomain is important for p300 to maintain the basal level of histone acetylation and to induce the transcriptional activation of p300-dependent genes. Nevertheless, the requirement of bromodomain and histone acetylation in p300-dependent gene transcription is determined in a gene specific manner.
Materials and Methods
Cell culture and reagents.
HeLa and SiHa cells were obtained from American Type Culture Collection (ATCC) and maintained in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% of Fetal Bovine Serum (FBS, Hyclone) and 1% of MEM Non-Essential amino acids (Invitrogene). The cells were cultured at 37°C in humidified air containing 5% CO2 for a minimum of 16 h prior to different treatments. Sodium butyrate, valproic acid and TSA were obtained from Sigma-Aldrich. Specific antibodies for p300, p21, Egr1 and E2F1 were obtained from Santa Cruz biotechnology. Anti-Ac-H3 (K9/14) and anti-β-actin antibodies were purchased from Sigma-Aldrich.
Transfection and western analysis.
Transient transfections were performed with FuGene (Promega) following the manufacturer's recommendation. After transfection, the cells were cultured in the growth medium for 24 h and treated as indicated. Whole cell extracts were isolated by incubating the cells in WCE buffer containing 50 mM Tris-HCl (pH 7.6), 400 mM NaCl, 10% glycerol, 0.5 mM DTT, 0.5 mM PMSF and 0.1% NP-40 for 30 min at 4°C as previously described.Citation31 The protein concentration was determined by Bradford assay (Bio-Rad) with the use of bovine serum albumin as a standard. Equal amounts of proteins were separated on SDS-PAGE and subjected to western analysis. Quantification of the western results was performed by using Scion Image computer software (Scion Corporation).
Real-time RT-PCR analysis.
Total RNA was isolated by using RNeasy Mini kit (Qiagen). Multiskan Spectrum spectrophotometer (Thermo) was used to determine the concentrations and the quality of total RNA. Equal amounts of RNA were used in reverse transcription with High Capacity cDNA Archive Kit—Multiscribe Reverse Transcriptase (Applied Biosystems). Quantitative real time PCR amplification was carried by using the Applied Biosystems 7500 Fast Real-Time PCR System. The levels of gene transcripts were quantified by using the TaqMan® reporter assay. Specific gene primers used in the amplification were pre-designed (TaqMan® primers by Applied Biosystems). 18S rRNA was used as a reference control.Citation23 The amount of targets, normalized to the 18S endogenous reference and relative to calibrator control is calculated using the arithmetic formula 2−ΔΔCT.
Pulse-chase.
Cells were cultured in DMEM supplemented with 10% of FBS for overnight and the media was replaced with methionine-free media. The cells were first pulsed for 2 h with 100 µCi/ml of 35S-methionine and chased for 2–24 h in regular medium.Citation44 The cells were then harvested for the preparation of whole cell extracts. For immunoprecipitation, the whole cell extracts were diluted to adjust the concentrations of NaCl to 150 mM by dilution buffer (10% glycerol, 20 mM Tris-HCl pH 8.0, 0.5 EDTA pH 8.0, 1 mM DTT, 1 mM PMSF and 1 mg/ml of BSA). The endogenous wild-type or mutant p300 protein was immunoprecipitated with a p300 specific antibody and washed three times with washing buffer (10% glycerol, 100 mM NaCl, 20 mM Tris-HCl pH 8.0, 0.5 mM EDTA pH 8.0, 1 mM DTT, 1 mM PMSF, 0.1% NP-40). The precipitates were then separated by SDS-PAGE and quantified by PhosphoImager.
Immunofluorescence microscopy.
Cells were grown on coverslips for about 16 h before treatment, fixed in paraformaldehyde, permeabilized in 0.2% NP-40 and blocked in PBS containing 0.2% tween 20 and 10% non-fat milk.Citation45 The coverslips were incubated with primary antibody specific for p300, then with rhodamine-conjugated secondary antibody, mounted in 90% glycerol and visualized by fluorescence microscopy.
Figures and Tables
Figure 1 Characterization of the wild-type and bromo-deficient p300 in HeLa and SiHa cells. (A) Equal amounts of whole cell extracts (50 µg) from the HeLa or SiHa cells were subjected to western analysis for the wild-type and bromo-deficient p300. The blots were then stripped and reprobed for β-actin as a loading control. (B) Quantification of the western blots was plotted as fold difference in reference to the wild-type p300 in the HeLa cells. Error bars represent the standard deviation of three independent experiments. Statistical significance is denoted by * to indicate p < 0.05 compared with the wild-type p300. (C) Relative mRNA level of bromo-deficient p300 in the SiHa cells was analyzed by quantitative real-time RT-PCR and presented as fold variation to the wild-type p300 transcripts in the HeLa cells. Error bars represent standard deviations of three independent experiments in duplicate samples. (D) Relative synthesis of the bromo-deficient p300 protein in the SiHa cells was determined following 2 h of pulse-labeling with 35S-methionine and expressed as fold variation of the wild-type p300 in the HeLa cells. Error bars represent standard deviations of three independent experiments. (E) The HeLa cells were pulse labeled with 35S-methionine for 2 h and chased for 6–24 h. The apparent half-life of the wild-type p300 protein was derived from three independent experiments in duplicate samples. (F) The experimental procedure was as in (E) except that the SiHa cells were used to determine the apparent half-life of the bromo-deficient p300. (G) Intracellular localization of the wild-type or bromodeficient p300 protein in the HeLa or SiHa cells was determined by immunofluorescence microscopy.
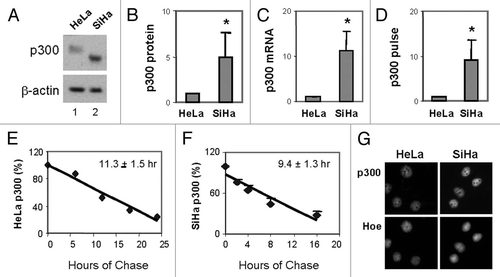
Figure 2 Effects of HDAC inhibitors on the expression of p300-dependent genes. (A) HeLa cells were treated with butyrate (NaB, 5 mM), valproic acid (VPA, 5 mM) or TSA (200 nM) for periods of 4, 8 or 16 h. Equal amounts of whole cell extracts (50 µg) were subjected to western analysis of p21, Egr1 or E2F1 protein. The blots were stripped and reprobed for β-actin as loading controls for quantification. (B) Quantification of the western blots was plotted as fold difference in reference to the untreated controls. (C and D) SiHa cells were treated and subjected to western analysis as for the HeLa cells in (A and B).
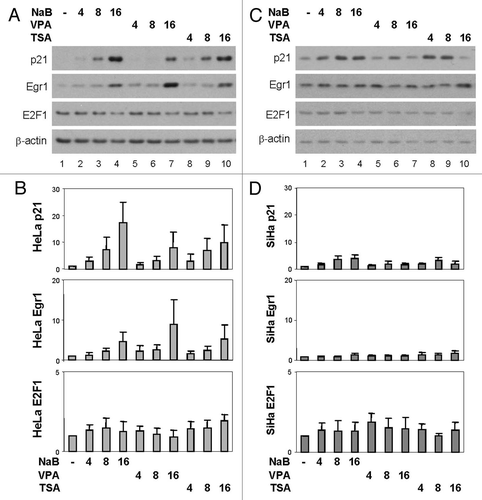
Figure 3 Effects of HDAC inhibitors on the transcription of p300-dependent genes. (A) Total RNA was extracted from the HeLa cells following 4 or 16 h treatments with butyrate (NaB, 5 mM), valproic acid (VPA, 5 mM) or TSA (200 nM) and reverse transcribed. The relative mRNA levels of p21 or Egr1 were determined by quantitative real time RT-PCR. 18S rRNA was used as an internal control. Values correspond to fold difference in reference to the untreated controls. Error bars present standard deviations of three independent experiments in duplicates. (B) The experimental procedure was as in (A) except that the SiHa cells were used for the real time RT-PCR analysis.
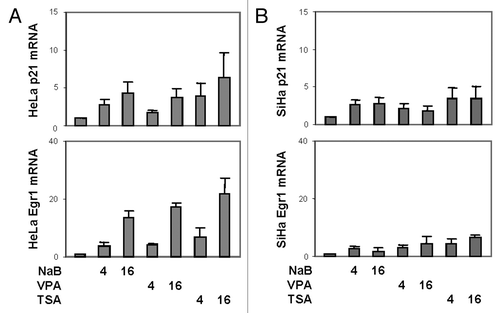
Figure 4 Global histone acetylation and p300-dependent gene expression. (A) Western analysis was performed by using specific antibody against acetylated histone 3 at K9/14 residue following 4 h of treatments of the HeLa or SiHa cells with butyrate (NaB, 5 mM), valproic acid (VPA, 5 mM) or TSA (200 nM). The blots were stripped and reprobed for β-actin as a loading control. (B) Quantification of the western blots is plotted as fold difference in reference to the untreated control of the HeLa cells. Error bars represent standard deviations of three independent experiments. Statistical significance is denoted by * to indicate p < 0.05 compared with the untreated control of HeLa cells. (C) The SiHa cells were transfected with expression plasmid (3 µg) for the full length p300, treated with butyrate, valproic acid or TSA for 8 h and then subjected to western analysis of p21 and Egr1. The blots were stripped and reprobed for β-tubulin as a loading control.
Acknowledgements
We thank Ms. Blaire Langlais for excellent technical assistance on the real time RT-PCR analysis of p300-depedent genes. This work was sponsored by operating grant from Canadian Institutes of Health Research (CIHR). During the course of this study, Q.L. was supported by an Investigator Award from the CIHR. F.M.G. is a recipient of Canada Graduate Study Master's Award from the CIHR.
References
- Arany Z, Newsome D, Oldread E, Livingston DM, Eckner R. A family of transcriptional adaptor proteins targeted by the E1A oncoprotein. Nature 1995; 374:81 - 84
- Lundblad JR, Kwok RP, Laurance ME, Harter ML, Goodman RH. Adenoviral E1A-associated protein p300 as a functional homologue of the transcriptional co-activator CBP. Nature 1995; 374:85 - 88
- Muraoka M, Konishi M, Kikuchi-Yanoshita R, Tanaka K, Shitara N, et al. p300 gene alterations in colorectal and gastric carcinomas. Oncogene 1996; 12:1565 - 1569
- Gayther SA, Batley SJ, Linger L, Bannister A, Thorpe K, Chin SF, et al. Mutations truncating the EP300 acetylase in human cancers. Nat Genet 2000; 24:300 - 303
- Suganuma T, Kawabata M, Ohshima T, Ikeda MA. Growth suppression of human carcinoma cells by reintroduction of the p300 coactivator. Proc Natl Acad Sci USA 2002; 99:13073 - 13078
- Petrij F, Giles RH, Dauwerse HG, Saris JJ, Hennekam RC, Masuno M, et al. Rubinstein-Taybi syndrome caused by mutations in the transcriptional co-activator CBP. Nature 1995; 376:348 - 351
- Miller RW, Rubinstein JH. Tumors in Rubinstein-Taybi syndrome. Am J Med Genet 1995; 56:112 - 115
- Roelfsema JH, White SJ, Ariyurek Y, Bartholdi D, Niedrist D, Papadia F, et al. Genetic Heterogeneity in Rubinstein-Taybi Syndrome: Mutations in Both the CBP and EP300 Genes Cause Disease. Am J Hum Genet 2005; 76
- Eckner R, Ludlow JW, Lill NL, Oldread E, Arany Z, Modjtahedi N, et al. Association of p300 and CBP with simian virus 40 large T antigen. Mol Cell Biol 1996; 16:3454 - 3464
- Patel D, Huang SM, Baglia LA, McCance DJ. The E6 protein of human papillomavirus type 16 binds to and inhibits co-activation by CBP and p300. EMBO J 1999; 18:5061 - 5072
- Eckner R, Ewen ME, Newsome D, Gerdes M, DeCaprio JA, Lawrence JB, et al. Molecular cloning and functional analysis of the adenovirus E1A-associated 300-kD protein (p300) reveals a protein with properties of a transcriptional adaptor. Genes Dev 1994; 8:869 - 884
- Goodman RH, Smolik S. CBP/p300 in cell growth, transformation and development. Genes Dev 2000; 14:1553 - 1577
- Jeanmougin F, Wurtz JM, Le Douarin B, Chambon P, Losson R. The bromodomain revisited. Trends Biochem Sci 1997; 22:151 - 153
- Marmorstein R, Berger SL. Structure and function of bromodomains in chromatin-regulating complexes. Gene 2001; 272:1 - 9
- Kraus WL, Manning ET, Kadonaga JT. Biochemical analysis of distinct activation functions in p300 that enhance transcription initiation with chromatin templates. Mol Cell Biol 1999; 19:8123 - 8135
- Syntichaki P, Topalidou I, Thireos G. The Gcn5 bromodomain co-ordinates nucleosome remodelling. Nature 2000; 404:414 - 417
- Dhalluin C, Carlson JE, Zeng L, He C, Aggarwal AK, Zhou MM. Structure and ligand of a histone acetyltransferase bromodomain. Nature 1999; 399:491 - 496
- Mujtaba S, He Y, Zeng L, Yan S, Plotnikova O, Sachchidanand, et al. Structural mechanism of the bromodomain of the coactivator CBP in p53 transcriptional activation. Mol Cell 2004; 13:251 - 263
- Mujtaba S, He Y, Zeng L, Farooq A, Carlson JE, Ott M, et al. Structural basis of lysine-acetylated HIV-1 Tat recognition by PCAF bromodomain. Mol Cell 2002; 9:575 - 586
- Owen DJ, Ornaghi P, Yang JC, Lowe N, Evans PR, Ballario P, et al. The structural basis for the recognition of acetylated histone H4 by the bromodomain of histone acetyltransferase gcn5p. EMBO J 2000; 19:6141 - 6149
- Jacobson RH, Ladurner AG, King DS, Tjian R. Structure and function of a human TAFII250 double bromodomain module. Science 2000; 288:1422 - 1425
- St-Germain JR, Chen J, Li Q. Involvement of PML nuclear bodies in CBP degradation through the ubiquitin-proteasome pathway. Epigenetics 2008; 3:342 - 349
- Chen J, Ghazawi FM, Bakkar W, Li Q. Valproic acid and butyrate induce apoptosis in human cancer cells through inhibition of gene expression of Akt/protein kinase B. Mol Cancer 2006; 5:71
- Johnstone RW. Histone-deacetylase inhibitors: novel drugs for the treatment of cancer. Nat Rev Drug Discov 2002; 1:287 - 299
- Marks PA, Jiang X. Histone deacetylase inhibitors in programmed cell death and cancer therapy. Cell Cycle 2005; 4:549 - 551
- Riggs MG, Whittaker RG, Neumann JR, Ingram VM. n-Butyrate causes histone modification in HeLa and Friend erythroleukaemia cells. Nature 1977; 268:462 - 464
- Phiel CJ, Zhang F, Huang EY, Guenther MG, Lazar MA, Klein PS. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer and teratogen. J Biol Chem 2001; 276:36734 - 36741
- Frew AJ, Johnstone RW, Bolden JE. Enhancing the apoptotic and therapeutic effects of HDAC inhibitors. Cancer Lett 2009; 280:125 - 133
- Johannessen CU, Johannessen SI. Valproate: past, present and future. CNS Drug Rev 2003; 9:199 - 216
- Yoshida M, Horinouchi S, Beppu T. Trichostatin A and trapoxin: novel chemical probes for the role of histone acetylation in chromatin structure and function. Bioessays 1995; 17:423 - 430
- Chen J, Halappanavar SS, St-Germain JR, Tsang BK, Li Q. Role of Akt/protein kinase B in the activity of transcriptional coactivator p300. Cell Mol Life Sci 2004; 61:1675 - 1683
- Avantaggiati ML, Carbone M, Graessmann A, Nakatani Y, Howard B, Levine AS. The SV40 large T antigen and adenovirus E1a oncoproteins interact with distinct isoforms of the transcriptional co-activator, p300. EMBO J 1996; 15:2236 - 2248
- Yaciuk P, Moran E. Analysis with specific polyclonal antiserum indicates that the E1A-associated 300-kDa product is a stable nuclear phosphoprotein that undergoes cell cycle phase-specific modification. Mol Cell Biol 1991; 11:5389 - 5397
- Smith JL, Freebern WJ, Collins I, De Siervi A, Montano I, Haggerty CM, et al. Kinetic profiles of p300 occupancy in vivo predict common features of promoter structure and coactivator recruitment. Proc Natl Acad Sci USA 2004; 101:11554 - 11559
- Finzer P, Kuntzen C, Soto U, zur Hausen H, Rosl F. Inhibitors of histone deacetylase arrest cell cycle and induce apoptosis in cervical carcinoma cells circumventing human papillomavirus oncogene expression. Oncogene 2001; 20:4768 - 4776
- Brownell JE, Allis CD. Special HATs for special occasions: linking histone acetylation to chromatin assembly and gene activation. Curr Opin Genet Dev 1996; 6:176 - 184
- Glaser KB, Staver MJ, Waring JF, Stender J, Ulrich RG, Davidsen SK. Gene expression profiling of multiple histone deacetylase (HDAC) inhibitors: defining a common gene set produced by HDAC inhibition in T24 and MDA carcinoma cell lines. Mol Cancer Ther 2003; 2:151 - 163
- Takai N, Desmond JC, Kumagai T, Gui D, Said JW, Whittaker S, et al. Histone deacetylase inhibitors have a profound antigrowth activity in endometrial cancer cells. Clin Cancer Res 2004; 10:1141 - 1149
- Ohshima T, Suganuma T, Ikeda M. A novel mutation lacking the bromodomain of the transcriptional coactivator p300 in the SiHa cervical carcinoma cell line. Biochem Biophys Res Commun 2001; 281:569 - 575
- Manning ET, Ikehara T, Ito T, Kadonaga JT, Kraus WL. p300 forms a stable, template-committed complex with chromatin: role for the bromodomain. Mol Cell Biol 2001; 21:3876 - 3887
- Agalioti T, Chen G, Thanos D. Deciphering the transcriptional histone acetylation code for a human gene. Cell 2002; 111:381 - 392
- Kraus WL, Kadonaga JT. p300 and estrogen receptor cooperatively activate transcription via differential enhancement of initiation and reinitiation. Genes Dev 1998; 12:331 - 342
- Zeng L, Zhou MM. Bromodomain: an acetyl-lysine binding domain. FEBS Lett 2002; 513:124 - 128
- Chen J, St-Germain JR, Li Q. B56 Regulatory Subunit of Protein Phosphatase 2A Mediates Valproic Acid-Induced p300 Degradation. Mol Cell Biol 2005; 25:525 - 532
- Chen J, Halappanavar S, Th' ng JP, Li Q. Ubiquitin-dependent distribution of the transcriptional coactivator p300 in cytoplasmic inclusion bodies. Epigenetics 2007; 2:92 - 99