
Abstract
Promoter methylation analysis of genes frequently silenced in breast cancer is a promising indicator of breast cancer risk, as these methylation events are thought to occur long before presentation of disease. The numerous exfoliated epithelial cells present in breast milk may provide the breast epithelial DNA needed for detailed methylation analysis and assessment of breast cancer risk. Fresh breast milk samples and health, lifestyle, and reproductive history questionnaires were collected from 111 women. Pyrosequencing analysis was conducted on DNA isolated from the exfoliated epithelial cells immunomagnetically separated from the total cell population in the breast milk of 102 women. A total of 65 CpG sites were examined in six tumor suppressor genes: PYCARD (also known as ASC or TMS1), CDH1, GSTP1, RBP1 (also known as CRBP1), SFRP1, and RASSF1. A sufficient quantity of DNA was obtained for meaningful analysis of promoter methylation; women donated an average of 86 ml of milk with a mean yield of 32,700 epithelial cells per ml. Methylation scores were in general low as expected of benign tissue, but analysis of outlier methylation scores revealed a significant relationship between breast cancer risk, as indicated by previous biopsy, and methylation score for several CpG sites in CDH1, GSTP1, SFRP1, and RBP1. Methylation of RASSF1 was positively correlated with women’s age irrespective of her reproductive history. Promoter methylation patterns in DNA from breast milk epithelial cells can likely be used to assess breast cancer risk. Additional studies of women at high breast cancer risk are warranted.
Acknowledgements
The authors thank the Avon Foundation for their generous support.
Figures and Tables
Figure 1 CpG sites examined in the six genes. The vertical lines represent individual CpG sites within the CpG island of the promoter region. The boxed areas enclose the sites analyzed in each assay. The transcriptional start site is marked with an arrow and the numbers in parentheses define the boxed region relative to the transcriptional start site.
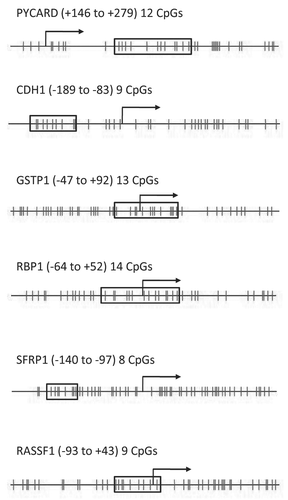
Figure 2 Participant demographics and sample yields from 102 of the 111 women who provided a breast milk sample.
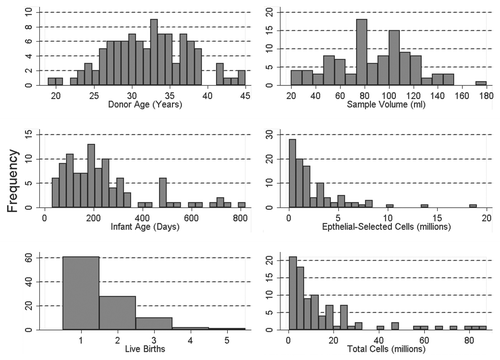
Figure 3 Comparison of pyrosequencing results obtained from methylated and non-methylated DNA for nine CpG sites spanning the transcriptional start site in the tumor suppressor gene RASS F1. (A) Pyrograms of methylated (top) and non-methylated (bottom) HMEC DNA show the nucleotide dispensation order on the x-axis, the signal intensity on the y-axis, the sequence being analyzed on the top and the percent methylated (gray columns) for each of the nine CpG sites. The narrower tan-shaded column at dispensation 18 marks a bisulfite-treatment control; as a non-methylated cytosine in the genomic DNA (a cytosine not followed by a guanine) will be completely converted to uracil with bisulfite treatment and then replaced with thymidine during the PCR. The dispensation begins with the addition of enzyme (E) followed by substrate (S) and then the dinucleotides. When no deviation from the predicted sequence is encountered and the bisulfite-conversion is ≥96% complete, the methylation score (%) is framed in a blue square at the top of the gray column and considered a perfect call. Small deviations from the expected sequence and/or bisulfite conversions between 92.5 and 95.5% complete are framed in yellow squares and the sequencing results are manually checked before accepting or rejecting the methylation score. Large deviations and/or bisulfite conversions less than 92.5% complete are framed in red squares (none shown) and the methylation score is rejected. (B) Pyrosequencing results from three separate PCR reactions of bisulfite-modified methylated control DNA (top three lines) and non-methylated control DNA (bottom three lines).
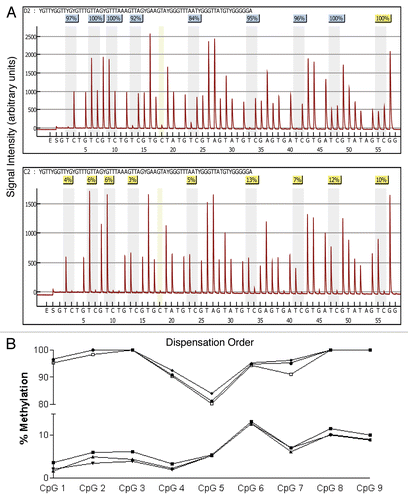
Figure 4 Box and whisker plots of percent overall mean methylation for each of the six genes analyzed in epithelial cell DNA isolated from breast milk. The filled circles represent outliers as defined in the methods. The top whisker represents the highest non-outlier score and the bottom whisker represents the lowest non-outlier score. The top line of the box represents the third quartile, the bottom line of the box the first quartile and the middle line the median. Samples sizes range between 93 and 102 as shown in .
Figure 5 Relationship between mother's age, age at first birth and percent overall mean methylation in RASSF1 epithelial cell DNA isolated from the breast milk of 102 women. The percent overall mean methylation for RASSF1 in women who first gave birth after they turned 25 years of age is represented as “O” (older at first birth) and the percent overall mean methylation for RASSF1 in women who first gave birth before they turned 25 years of age is represented as “Y” (younger at first birth).
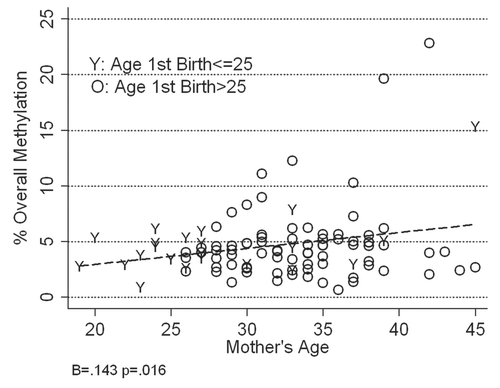
Figure 6 Relationship between mother's age and percent methylation for each of the nine CpG sites analyzed in RASSF1 epithelial cell DNA isolated from the breast milk of 102 women.
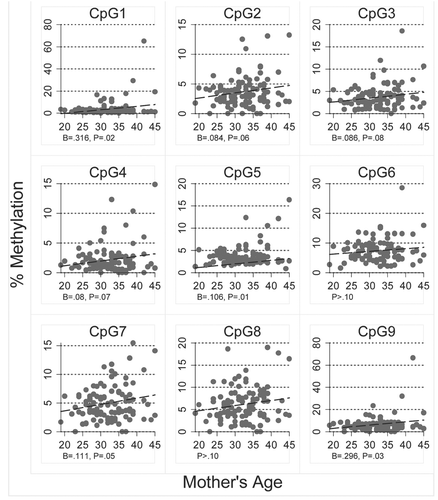
Table 1 Mean percent methylated DNA for each of the six genes examined