Abstract
Global DNA hypomethylation affecting repeat sequences has been reported in different cancer types. Herein, we investigated the methylation levels of repetitive DNA elements in chronic lymphocytic leukemia (CLL), their correlation with the major cytogenetic and molecular features, and clinical relevance in predicting therapy-free survival (TFS). A quantitative bisulfite-PCR Pyrosequencing method was used to evaluate methylation of Alu, long interspersed nuclear elements-1 (LINE-1) and satellite-α (SAT-α) sequences in 77 untreated early-stage (Binet A) CLL patients. Peripheral B-cells from 7 healthy donors were used as controls. Methylation levels (median %5mC) were lower in B-CLLs compared with controls (21.4 vs. 25.9; 66.8 vs. 85.7; 84.0, vs. 88.2 for Alu, LINE-1 and SAT-α, respectively) (p < 0.001). Among CLL patients, a significant association was observed with 17p13.1 deletion (16.8 vs. 22.4; 51.2 vs. 68.5; 52.6 vs. 85.0, for Alu, LINE-1 and SAT-α) but not with other major genetic lesions, IgVH mutation status, CD38 or ZAP-70 expression. Follow-up analyses showed that lower SAT-α methylation levels appeared to be an independent prognostic marker significantly associated with shorter TFS. Our study extended previous limited evidences in methylation of repetitive sequences in CLL suggesting an important biological and clinical relevance in the disease.
Introduction
Chronic lymphocytic leukemia (CLL) is a clinically heterogeneous disorder characterized by a disparate clinical course with survival varying from months to decades.Citation1 Several biomarkers have been correlated with aggressive disease, including chromosomal abnormalities such as deletions at chromosomes 11q22.3 and 17p13.1,Citation2 the mutation status of the variable regions of the heavy chain immunoglobulin genes (IgVH),Citation3 the expression of the 70 kd zeta-chain T-cell receptor-associated protein kinase (ZAP-70) and CD38 cell surface antigen.Citation4,Citation5 However, patients with a predicted good prognosis may show an aggressive disease, suggesting that stratification according to these parameters does not fully explain the clinical heterogeneity of CLL.
DNA methylation, one of the most investigated epigenetic mechanisms in mammals, is a reversible event that modifies genome function and chromosomal stability through the addition of methyl groups to cytosine to form 5-Methyl-Cytosine (5mC) mostly occurring in a 5'-CpG-3' sequence context.Citation6 CpG dinucleotides are often aberrantly methylated in human cancers to give an overall reduction of 5mC (global DNA hypomethylation) despite regional hypermethylation at some CpG islands.Citation7 The enzymes responsible for CpG methylation are the DNA methyltransferases (DNMTs) including DNMT1, DNMT3a and DNMT3b. DNMT1 is known to be the “maintenance” enzyme whereas both DNMT3a and DNMT3b are known as de novo methyltransferases.Citation8,Citation9
Hypomethylation of repetitive elements (i.e., transposable elements, satellite DNA, LINEs and SINEs) is thought to largely account for the global hypomethylation commonly observed in human cancers.Citation10,Citation11 A significant fraction of these elements is represented by interspersed sequences such as Alu, accounting for ~10% of the human genome and LINE-1.Citation10 Satellite DNA sequences (i.e., Sat-α) are generally found in centromeres or centromere-adjacent heterochromatin.Citation12 Repetitive elements, mainly the interspersed repeated DNA sequences, contain numerous CpG dinucleotides; therefore, the methylation status of these sequences is relevant to the understanding of the global DNA methylation pattern.Citation13
Global or gene-specific hypomethylation,Citation14-Citation17 as well as hypermethylation affecting putative tumor-suppressor genes,Citation18-Citation22 have been reported in CLL. Despite previous studies describing global DNA hypomethylation in CLL, the role of specific repetitive elements has been poorly investigated.Citation23 The relationship of global DNA methylation levels with clinical and biological risk factors of the disease, as well as its clinical course, remain uncertain.
The goal of the present work was to investigate the methylation levels of Alu, LINE-1 and SAT-α repetitive sequences and correlate them with the major biological and cytogenetic markers known to predict clinical outcome in CLL. The association of repetitive DNA sequences methylation with disease progression, measured as the time elapsed from diagnosis to first treatment (therapy-free survival, TFS), was also investigated. To this end a quantitative PCR-Pyrosequencing approach was used to determine the levels of repetitive sequences methylation in a panel of highly purified (>90%) peripheral mononuclear CD19+ cells from seven healthy donors and 77 untreated CLLs in early stage disease (Binet stage A).
Results
Molecular characterization of the CLL patients.
The molecular and biological characteristics of the 77 patients included in the study are shown in . ZAP-70, CD38 and IgVH mutational analyses were performed in 74, 71 and 76 cases, respectively. Forty-eight patients had un-mutated IgVH genes; ZAP-70 and CD38 were positive in 28 and 35 cases, respectively; 13q14.3 deletion was present as a sole abnormality in 21/33 patients while in the remaining cases it was combined with 17p13.1 (n = 4) or 11q22.3 deletions (n = 7) or both (n = 1). The 11q22.3 and 17p13.1 deletions were detected as sole abnormality in 6 and 7 patients, respectively. Trisomy 12 occurred in 17 patients, as a sole abnormality in all cases.
Table 1. Biological and molecular features of CLL patients included in the study
Methylation levels in healthy subjects and CLL samples.
The methylation levels of Alu, LINE-1 and SAT-α DNA in B-CLLs were compared with value obtained for CD19+ purified peripheral B cells from seven healthy donors (). CLLs showed a statistically significant decrease of Alu (median: 21.4%5mC), LINE-1 (66.8%5mC) and SAT-α DNA (84.0%5mC) methylation levels as compared with controls (25.9%5mC, 85.7%5mC and 88.2%5mC, respectively) (p < 0.001).
Table 2. Global DNA methylation levels in healthy subjects compared with CLL patients
Correlation of clinically relevant parameters with the DNA methylation levels.
We evaluated global DNA methylation levels of repetitive sequences in the context of different cytogenetic groups. A statistically significant association was only found between Alu, LINE-1 or SAT-α hypomethylation levels and the occurrence of the 17p13 deletion (p = 1.107e-04 for Alu; 1.535e-05 for LINE-1 and 3.88e-05 for SAT-α), ().
Figure 1. DNA methylation levels in relation to different cytogenetic groups. Box plot representation of DNA methylation levels. The p values corresponding to each methylation marker are shown.
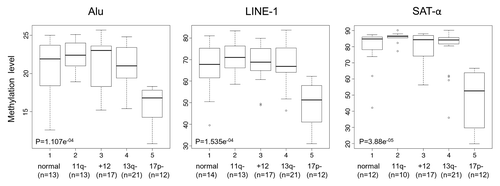
The median %5mC of 17p13 deleted patients vs. those without this lesion were 16.8 vs. 22.4 for Alu, 51.2 vs. 68.5 for LINE-1 and 52.6 vs. 85.0 for SAT-α, respectively. We then extended correlation analysis to other prognostic parameters such as IgVH mutation status, CD38 or ZAP-70 expression, but no significant associations with DNA methylation levels were observed ().
Table 3. DNA methylation levels related to different biological markers in CLL patients
DNMT1, DNMT3a and DNMT3b expression in healthy subjects and CLL patients.
To investigate the relationship between DNA hypomethylation and DNMTs expression we evaluated the absolute expression levels of three distinct DNMT genes in 42 out of 77 CLLs and 6 healthy donors available in our gene expression profiling database. DNMT1 showed a significantly decreased absolute median expression in CLLs (median: 103.8, range: 52.9–194.4) as compared with normal B cells (median: 149, range: 107.4–212.7, p = 0.0084), whereas no significant differences were observed for DNMT3a and DNMT3b mRNA expression ().
Figure 2. Boxplot representation of absolute RNA expression levels of the three DNMTs(DNMT1, DNMT3a and DNMT3b) in healthy subjects and CLL patients as assessed by microarray analysis. A significantly decreased absolute median RNA expression level in CLLs vs. healthy donors was found for DNMT1 (p = 0.0084).

Correlation between DNA methylation markers.
A moderate correlation was found between the methylation levels of Alu and LINE-1, Alu and SAT-α DNA and LINE-1 and SAT-α (| = 0.33, 0.52 and 0.36, respectively; p < 0.001). We did not observe significant association between the DNA methylation of Alu, LINE-1 and SAT-α DNA and expression levels of the three DNMTs (data not shown).
Prognostic value of Alu, LINE-1 and SAT-α methylation levels.
Clinical data were available for 65 patients. The median follow-up was two years (range 1–10 years) and 39 patients had received treatment by the end of the study. As expected, univariate Cox analysis showed that the patients who were CD38 positive (n = 32) (HR = 2.0, 95% C.I. 1.1–4.0, p = 0.032), ZAP-70 positive (n = 25) (HR = 1.9, 95% C.I. 1.1–3.8, p = 0.046), IgVH un-mutated (n = 40) (HR = 2.4, 95% C.I. 1.1–5.1, p = 0.022) and cases deleted for 17p13.1 (n = 5) (HR = 4.1, 95% C.I. 1.4–12.3, p < 0.0001) were at higher risk of starting treatment.
In order to examine the potential prognostic relevance of hypomethylation of each marker, CLL patients were tested for the methylation levels of Alu, LINE-1 and SAT-α which maximize the risk of therapy. The analysis of each methylation marker revealed a significantly higher risk of starting treatment for patients with lower Alu (n = 5, cut-off = 15.3%5mC, HR = 3.7, 95% C.I. 1.4–9.8, p = 0.008) and SAT-α (n = 13, cut-off = 69.0%5mC, HR = 4.9, 95% C.I. 2.2–10.8, p < 0.0001) methylation levels (), whereas no statistically significant association was demonstrated with regard to LINE-1. Notably, after correction for multiple testing comparison, only SAT-α retained a significant correlation with TFS (p-adj = 0.0032, HR = 4.7, 95% C.I. 1.7–13). Finally, the methylation status of SAT-α adjusted for CD38, ZAP-70 expression, IgVH mutational status or 17p13.1 deletion indicated the independent prognostic power of SAT-α hypomethylation in relation with the risk of treatment start (). In these models, CD38, ZAP-70, IgVH mutational status and 17p13.1 deletion were no longer statistically significant.
Figure 3. Cox-derived estimated curves according to the SAT-α methylation levels. The curves show the proportional hazard ratio estimate according to SAT-α methylation level below (n = 13, group A) or above (n = 47, group B) the cut-off value (69.0%5mC).
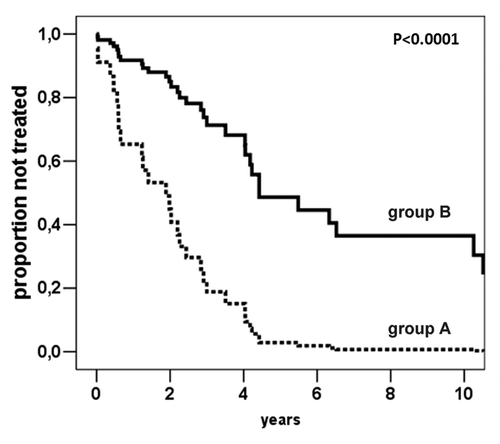
Table 4. Multivariate Cox proportional hazards analysis of Sat-α methylation level along with either CD38 or ZAP-70 expression or IgVH mutational status as predictors of TFS
Discussion
Many studies have focused on the relevance and clinical applications of DNA hypermethylation in human cancer.Citation24 However, hypomethylation of the genome affecting intergenic and intronic DNA regions, such as repeat sequences and transposable elements, is believed to result in chromosomal instability, increased mutation events and altered gene expression.Citation25,Citation26
Although a reduced overall 5-Methyl-Cytosine (5mC) content has previously been demonstrated in CLL,Citation14,Citation15 the role of repetitive element methylation has been scarcely investigated in this disease.Citation23 In the current study, we quantified the methylation levels of Alu, LINE-1 and SAT-α repetitive sequences in a panel of seven healthy donors and 77 untreated B-CLLs in early stage disease, using the Pyrosequencing technology. This approach is widely thought to have a greater precision, accuracy, sensitivity and reproducibility than the capillary electrophoresis or methylation-sensitive restriction enzymes methods generally used in previous studies dealing with global genomic methylation.Citation13,Citation27 Finally, to the best of our knowledge, this is the first comprehensive report describing global DNA methylation patterns of repeated elements in CLL and their relationship with clinically relevant biological and cytogenetic markers, as well as their potential prognostic value in terms of TFS.
The significantly decreased methylation levels of Alu, LINE-1 and SAT-α repetitive elements found in CLL samples when compared with healthy donors extended previous evidence showing that global hypomethylation is a common feature of CLLs, a finding recently supported by high-resolution methylation micro-array analysis of a limited number of patients.Citation28 Since our series involved a panel of untreated CLL patients in early stage disease, this finding strongly suggest that hypomethylation of repetitive elements may represent an early event in CLL similar to that observed in other tumors.Citation29,Citation30
Notably, our data indicate that SAT-α hypomethylation may represent an independent negative prognostic marker that significantly correlates with the need of starting treatment at the identified cut-off below 70%5mC. This finding suggests that global DNA hypomethylation may contribute to CLL tumor progression, likely promoting genomic instability as a consequence of destabilization of repetitive sequences.Citation31,Citation32 Hypomethylation of DNA satellite repeats in pericentromeric regions has been shown to lead to chromatin decondensation and consequent chromosomal fragility. The high frequency of chromosome rearrangements/breakpoints found in different cancer types and associated with tumor progression has been correlated with a marked role of satellite repeats hypomethylation.Citation33 Finally, it should be noted that the hypomethylation of the three analyzed repetitive sequences appeared to be correlated, albeit at moderate levels. This finding may suggest a common mechanism of demethylation of such elements in CLL as reported in other types of tumor.Citation34,Citation35
With regards to molecular and biological prognostic markers in CLL, of particular interest is the evidence of a significant hypomethylation of Alu, LINE-1 and SAT-α in CLLs carrying 17p13.1 deletion, which is known to be associated with an aggressive clinical course.Citation2 No significant correlation with other genetic lesions, CD38 or ZAP-70 expression and IgVH mutation status was observed. However, with regards to the IgVH mutation status, Lyko and colleagues,Citation14 using a different quantitative method and a chi-squared statistics, have earlier identified an estimated cut-off value in global cytosine methylation that maximized the differences in the distribution of IgVH mutated and un-mutated cases. The same statistics applied to our data set allowed to identify 23.8% (p = 0.0223) and 86.1% (p = 0.0107) as cut-off values for Alu and SAT-α methylation levels, respectively (data not shown). However, such values are irrespective of the global distribution of IgVH mutated/un-mutated cases, according to the methylation status (i.e., the medians are not significantly different) and may only account for the prevalence of IgVH un-mutated cases among those cases with lower methylation levels.
DNMTs enzymes are thought to play an important role in the physiological DNA methylation; however, their involvement in cancer remains to be fully clarified.Citation36 Notably, we observed significant changes only in DNMT1 mRNA levels in CLLs when compared with normal lymphocytes. However, we did not observe significant correlation between the expression levels of any of the three DNMTs investigated and DNA hypomethylation. A correlation was not even detectable between DNMTs expression level and specific cytogenetic subgroups, including patients with 17p deletion. Interestingly, it has been recently reported that increase of DNMT1 expression in lung cancer is mediated by p53 deficiency.Citation22 Our data suggest that DNMT1 expression level is independent of p53 deficiency in CLL tumors. Nevertheless, based on the limited number of cases, this finding warrants confirmation on larger series of patients.
The lack of correlation between DNMTs expression level and DNA hypomethylation suggests that other mechanisms, including interactions with specific chromatin components, transcription factors, replication and repair machinery, may be responsible for the observed genome-wide hypomethylation in CLL.Citation25 In contrast, using quantitative RT-PCR other investigators have reported a normal DNMT1 expression and significant reduction of DNMT3b in CLLs when compared with normal B cell.Citation37 These different results could be most likely due to the limited number and the higher prevalence of advanced stages characterizing the series of patients included in the previous study. Additionally, we have investigated the absolute expression levels of DNMTs probe sets in an independent, publicly available CLL microarray database (GSE6691)Citation38 profiled on Affymetrix GeneChip® HG-U133A arrays and including normal B cells. Differently from our data, we did not find any significant difference in DNMT1 expression levels between normal donors and CLL patients. However, the very limited number of CLL samples (11 cases) included in the GSE6691 database could probably affect the reproducibility of the analysis.
In summary, our study extended previous limited evidence of global hypomethylation of repetitive sequences in CLL demonstrating a significant correlation with specific cytogenetic risk groups, such as 17p deleted patients. In addition, it provided evidence that the hypomethylation of specific repetitive sequences may have clinical relevance in CLL and may be used as a novel prognostic indicator of unfavorable disease progression. Further studies on larger representative prospective series of CLLs are needed to fully confirm the present findings.
Materials and Methods
Patient samples.
Highly purified (>90%) peripheral mononuclear CD19+ cell populations from seven healthy donors (four males; median age 58 years, range 42–64) and 77 CLL untreated patients in early stage disease (Binet stage A) were included in this study. All the blood samples were collected within one year after diagnosis. Patients belonged to a database from a collaborative Italian study.Citation39 The inclusion criteria were a diagnosis of typical CLL based on morphological and phenotypical analyses (i.e., the co-expression of CD19, CD5 and CD 23 and weak SmIg, the monotypical expression of | or ⌊ light chains by neoplastic cells). Forty-six patients were males and 31 were females; the mean and median age at diagnosis was 64 and 65 years, respectively (range 27–87). In accordance with our institutional guidelines, all of the patients gave their informed consent. No conventional cytogenetic (G-banding) analyses were available. In order to reduce any selection bias as much as possible, patients were selected based on the distribution of cytogenetic lesions and IgVH mutation status. Forty-two out of 77 CLL patients and six out of seven healthy donors were previously profiled with the GeneChip® Human Genome U133A (HG-U133A) arrays and publicly available on the Gene Expression Omnibus (GEO) website under accession number GSE16746.
Sample preparation, immunophenotype and prognostic markers.
Peripheral blood mononuclear cells from CLL patients were isolated by Ficoll-Hypaque (Seromed, Biochrom KG, Berlin, Germany) density-gradient centrifugation. If CLL cells were less than 90%, T cells, NK cells and monocytes were removed by CD3, CD56, CD16 and CD14 monoclonal antibody (mAb) treatment (Becton Dickinson, & Co.) followed by magnetic beads (Goat Anti-Mouse IgG Dynabeads, Dynal Biotech ASA, Oslo, Norway).Citation40
The proportion of CD5/CD19/CD23 triple positive B cells in the suspension was determined by direct immunofluorescence performed using a FACS-sort flow cytometer (BD Biosciences, San José, CA) with antibodies to: CD19 FITC/PE, CD23 PE and CD5 Cy-Chrome (BD Biosciences). CD38-positive leukemic cells were measured by triple staining with CD19 FITC, CD38 PE and CD5 Cy-Chrome (Becton Dickinson & Co., Sunnyvale, CA). The cells were analyzed using a FACSCalibur flow cytometer (Becton Dickinson & Co.) as previously described.Citation40 ZAP-70 was determined by flow-cytometry with a ZAP-70 FITC (clone 2F3.2, Upstate, Lake Placid, NY) or an isotype control mAb (mouse IgG2a FITC Becton Dickinson).Citation39,Citation40 A 30% cutoff was used for both CD38 and ZAP-70 expression. IgVH gene usage and mutational status was determined as previously described and cutoff of 2% was used to distinguish mutated and un-mutated patients.
Interphase fluorescence in situ hybridization (I-FISH).
The 77 CLL patients were investigated by I-FISH for the most common genomic aberrations described in CLL, including trisomy 12 and chromosome deletion at 17p13.1, 11q22.3 and 13q14.3 loci. The FISH procedure and specific probes for chromosome aberrations detection have been previously described.Citation41
DNA extraction and bisulfite treatment of the DNA.
DNA was extracted by a commercial kit (Promega, Madison, WI). One microgram DNA (concentration 50 ng/∝l) was treated using EZ DNA Methylation-GoldTM Kit (Zymo Research, Orange, CA) according to the manufacturer’s protocol. Final elution was performed with 30 ∝l of M-Elution Buffer. Bisulfite-treated DNA was stored at -20°C for no more than one week and used shortly after treatment.
Bisulfite polymerase chain reaction and pyrosequencing.
Analysis of DNA methylation in Alu, LINE-1 and SAT-α repetitive element was performed by using Pyrosequencing and bisulfite-polymerase chain reaction (PCR).Citation13,Citation34,Citation42 In brief, the samples were bisulfite-treated and PCR-amplified. Pyrosequencing was performed using the PyroMark MD Pyrosequencing System (Pyrosequencing, Inc., Uppsala, Sweden). Methylation quantification was performed using the provided software. The degree of methylation was expressed as %5-methylated cytosines (%5mC) over the sum of methylated and unmethylated cytosines. All assays were run in duplicate to measure inter-run variability and reduce measurement error. The average of the three triplicates was used in the statistical analysis. Analytical variability [coefficient of variation (CV) calculated in duplicate runs] was 0.7% for the LINE-1, 1.6% for the Alu and 0.1% for the SAT-a.
Statistical analysis.
All data were statistically analyzed using conventional procedures in R (www.r-project.org) and SPSS statistical package for Windows, release 15.0, 2006 software (SPSS UK, Working, Surrey, UK). Kendall’s | correlation was used to assess associations among different DNA methylation variables. To test for differences among two distributions Wilcoxon Rank Sum Test was used and Kruskal-Wallis test if more than two groups of patients were compared. The prognostic impact of the biological and molecular variables was investigated by Cox proportional hazard models. The regression was evaluated in terms of Hazard Ratio (HR) with a cutoff for significance of p value <0.05. A custom R procedure was used to identify cut-off values for ALU, LINE or SAT-α methylation which maximized the HR of undergoing treatment, applying a Holm-Bonferroni correction for multiple testing comparisons.
Additional material
Download Zip (160.6 KB)Acknowledgements
This study was supported by grants from the Associazione Italiana Ricerca sul Cancro (IG-4659 to A.N., IG-6016 to A.B. and RG-6432 to F.M.); the Associazione Italiana Leucemie, Sezione Milano; Fondazione ‘Amelia Scorza’ ONLUS, Cosenza; Progetti Strategici-Ricerca Finalizzata Ministero Italiano della Salute (RFPS20063339960 to G.C. and RFPS2006340196 to F.M. and M.F.); the Fondo Investimento per la Ricerca di Base (RBIP06LCA9 to M.F.). L.A. is supported by a fellowship from the Fondazione Italiana Ricerca sul Cancro.
Note
Supplementary materials can be found at: www.landesbioscience.com/journals/epigenetics/article/13528
References
- Chiorazzi N, Rai KR, Ferrarini M. Chronic lymphocytic leukemia. N Engl J Med 2005; 352:804 - 15
- Dohner H, Stilgenbauer S, Benner A, Leupolt E, Krober A, Bullinger L, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med 2000; 343:1910 - 6
- Hamblin TJ, Davis Z, Gardiner A, Oscier DG, Stevenson FK. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood 1999; 94:1848 - 54
- Damle RN, Wasil T, Fais F, Ghiotto F, Valetto A, Allen SL, et al. Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. Blood 1999; 94:1840 - 7
- Wiestner A, Rosenwald A, Barry TS, Wright G, Davis RE, Henrickson SE, et al. ZAP-70 expression identifies a chronic lymphocytic leukemia subtype with unmutated immunoglobulin genes, inferior clinical outcome and distinct gene expression profile. Blood 2003; 101:4944 - 51
- Bird A. DNA methylation patterns and epigenetic memory. Genes Dev 2002; 16:6 - 21
- Feinberg AP, Gehrke CW, Kuo KC, Ehrlich M. Reduced genomic 5-methylcytosine content in human colonic neoplasia. Cancer Res 1988; 48:1159 - 61
- Robertson KD. it-Si-Ali S, Yokochi T, Wade PA, Jones PL, Wolffe AP. DNMT1 forms a complex with Rb, E2F1 and HDAC1 and represses transcription from E2F-responsive promoters. Nat Genet 2000; 25:338 - 42
- Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999; 99:247 - 57
- Ehrlich M. DNA methylation in cancer: too much, but also too little. Oncogene 2002; 21:5400 - 13
- Qu G, Dubeau L, Narayan A, Yu MC, Ehrlich M. Satellite DNA hypomethylation vs. overall genomic hypomethylation in ovarian epithelial tumors of different malignant potential. Mutat Res 1999; 423:91 - 101
- Lee M, Hadi M, Hallden G, Aponte GW. Peptide YY and neuropeptide Y induce villin expression, reduce adhesion and enhance migration in small intestinal cells through the regulation of CD63, matrix metalloproteinase-3 and Cdc42 activity. J Biol Chem 2005; 280:125 - 36
- Yang AS, Estecio MR, Doshi K, Kondo Y, Tajara EH, Issa JP. A simple method for estimating global DNA methylation using bisulfite PCR of repetitive DNA elements. Nucleic Acids Res 2004; 32:38
- Lyko F, Stach D, Brenner A, Stilgenbauer S, Dohner H, Wirtz M, et al. Quantitative analysis of DNA methylation in chronic lymphocytic leukemia patients. Electrophoresis 2004; 25:1530 - 5
- Stach D, Schmitz OJ, Stilgenbauer S, Benner A, Dohner H, Wiessler M, et al. Capillary electrophoretic analysis of genomic DNA methylation levels. Nucleic Acids Res 2003; 31:2
- Wahlfors J, Hiltunen H, Heinonen K, Hamalainen E, Alhonen L, Janne J. Genomic hypomethylation in human chronic lymphocytic leukemia. Blood 1992; 80:2074 - 80
- Hanada M, Delia D, Aiello A, Stadtmauer E, Reed JC. bcl-2 gene hypomethylation and high-level expression in B-cell chronic lymphocytic leukemia. Blood 1993; 82:1820 - 8
- Chim CS, Pang R, Liang R. Epigenetic dysregulation of the Wnt signalling pathway in chronic lymphocytic leukaemia. J Clin Pathol 2008; 61:1214 - 9
- Liu TH, Raval A, Chen SS, Matkovic JJ, Byrd JC, Plass C. CpG island methylation and expression of the secreted frizzled-related protein gene family in chronic lymphocytic leukemia. Cancer Res 2006; 66:653 - 8
- Rush LJ, Raval A, Funchain P, Johnson AJ, Smith L, Lucas DM, et al. Epigenetic profiling in chronic lymphocytic leukemia reveals novel methylation targets. Cancer Res 2004; 64:2424 - 33
- Seeliger B, Wilop S, Osieka R, Galm O, Jost E. CpG island methylation patterns in chronic lymphocytic leukemia. Leuk Lymphoma 2009; 50:419 - 26
- Tong WG, Wierda WG, Lin E, Kuang SQ, Bekele BN, Estrov Z, et al. Genome-wide DNA methylation profiling of chronic lymphocytic leukemia allows identification of epigenetically repressed molecular pathways with clinical impact. Epigenetics 2010; 5:499 - 508
- Dante R. nte-Paire J, Rigal D, Roizes G. Methylation patterns of long interspersed repeated DNA and alphoid repetitive DNA from human cell lines and tumors. Anticancer Res 1992; 12:559 - 63
- Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med 2003; 349:2042 - 54
- Wilson AS, Power BE, Molloy PL. DNA hypomethylation and human diseases. Biochim Biophys Acta 2007; 1775:138 - 62
- Pattamadilok J, Huapai N, Rattanatanyong P, Vasurattana A, Triratanachat S, Tresukosol D, et al. LINE-1 hypomethylation level as a potential prognostic factor for epithelial ovarian cancer. Int J Gynecol Cancer 2008; 18:711 - 7
- Aparicio A, North B, Barske L, Wang X, Bollati V, Weisenberger D, et al. LINE-1 methylation in plasma DNA as a biomarker of activity of DNA methylation inhibitors in patients with solid tumors. Epigenetics 2009; 4:176 - 84
- Kanduri M, Cahill N, Goransson H, Enstrom C, Ryan F, Isaksson A, et al. Differential genome-wide array-based methylation profiles in prognostic subsets of chronic lymphocytic leukemia. Blood 2009; 115:296 - 305
- Kondo Y, Issa JP. Epigenetic changes in colorectal cancer. Cancer Metastasis Rev 2004; 23:29 - 39
- Suter CM, Martin DI, Ward RL. Hypomethylation of L1 retrotransposons in colorectal cancer and adjacent normal tissue. Int J Colorectal Dis 2004; 19:95 - 101
- Costa FF, Paixao VA, Cavalher FP, Ribeiro KB, Cunha IW, Rinck JA Jr., et al. SATR-1 hypomethylation is a common and early event in breast cancer. Cancer Genet Cytogenet 2006; 165:135 - 43
- Itano O, Ueda M, Kikuchi K, Hashimoto O, Hayatsu S, Kawaguchi M, et al. Correlation of postoperative recurrence in hepatocellular carcinoma with demethylation of repetitive sequences. Oncogene 2002; 21:789 - 97
- Eden A, Gaudet F, Waghmare A, Jaenisch R. Chromosomal instability and tumors promoted by DNA hypomethylation. Science 2003; 300:455
- Bollati V, Fabris S, Pegoraro V, Ronchetti D, Mosca L, Deliliers GL, et al. Differential repetitive DNA methylation in multiple myeloma molecular subgroups. Carcinogenesis 2009; 30:1330 - 5
- Roman-Gomez J, Jimenez-Velasco A, Agirre X, Castillejo JA, Navarro G, San Jose-Eneriz E, et al. Repetitive DNA hypomethylation in the advanced phase of chronic myeloid leukemia. Leuk Res 2008; 32:487 - 90
- Weisenberger DJ, Velicescu M, Cheng JC, Gonzales FA, Liang G, Jones PA. Role of the DNA methyltransferase variant DNMT3b3 in DNA methylation. Mol Cancer Res 2004; 2:62 - 72
- Kn H, Bassal S, Tikellis C, El-Osta A. Expression analysis of the epigenetic methyltransferases and methyl-CpG binding protein families in the normal B-cell and B-cell chronic lymphocytic leukemia (CLL). Cancer Biol Ther 2004; 3:989 - 94
- Gutierrez NC, Ocio EM, de Las RJ, Maiso P, Delgado M, Ferminan E, et al. Gene expression profiling of B lymphocytes and plasma cells from Waldenstrom’s macroglobulinemia: comparison with expression patterns of the same cell counterparts from chronic lymphocytic leukemia, multiple myeloma and normal individuals. Leukemia 2007; 21:541 - 9
- Morabito F, Cutrona G, Gentile M, Matis S, Todoerti K, Colombo M, et al. Definition of progression risk based on combinations of cellular and molecular markers in patients with Binet stage A chronic lymphocytic leukaemia. Br J Haematol 2009; 146:44 - 53
- Cutrona G, Colombo M, Matis S, Fabbi M, Spriano M, Callea V, et al. Clonal heterogeneity in chronic lymphocytic leukemia cells: superior response to surface IgM cross-linking in CD38, ZAP-70-positive cells. Haematologica 2008; 93:413 - 22
- Fabris S, Mosca L, Todoerti K, Cutrona G, Lionetti M, Intini D, et al. Molecular and transcriptional characterization of 17p loss in B-cell chronic lymphocytic leukemia. Genes Chromosomes Cancer 2008; 47:781 - 93
- Bollati V, Baccarelli A, Hou L, Bonzini M, Fustinoni S, Cavallo D, et al. Changes in DNA methylation patterns in subjects exposed to low-dose benzene. Cancer Res 2007; 67:876 - 80