
Abstract
DNA methylation is one of the most important heritable epigenetic modifications of the genome and is involved in the regulation of many cellular processes. Aberrant DNA methylation has been frequently reported to influence gene expression and subsequently cause various human diseases, including cancer. Recent rapid advances in next-generation sequencing technologies have enabled investigators to profile genome methylation patterns at single-base resolution. Remarkably, more than 20 eukaryotic methylomes have been generated thus far, with a majority published since November 2009. Analysis of this vast amount of data has dramatically enriched our knowledge of biological function, conservation and divergence of DNA methylation in eukaryotes. Even so, many specific functions of DNA methylation and their underlying regulatory systems still remain unknown to us. Here, we briefly introduce current approaches for DNA methylation profiling and then systematically review the features of whole genome DNA methylation patterns in eight animals, six plants and five fungi. Our systematic comparison provides new insights into the conservation and divergence of DNA methylation in eukaryotes and their regulation of gene expression. This work aims to summarize the current state of available methylome data and features informatively.
Acknowledgements
We would like to thank Jeffery Ewers for critical reading and improving the manuscript. This work was supported by a NIH grant (R03LM009598) from the National Library of Medicine, Vanderbilt's Specialized Program of Research Excellence in GI Cancer grant (P50CA95103) and the VICC Cancer Center Core grant (P30CA68485).
Figures and Tables
Figure 1 Evolution of DNA methylation level of the available eukaryote methylomes. The phylogenetic tree was based on NCBI Taxonomy Browser (www.ncbi.nlm.nih.gov/taxonomy/taxonomyhome.html). Only the topology is shown and the branch lengths are not proportional to evolutionary divergence time. Green and red boxes indicate high methylation of gene body and transposon elements (TEs), respectively. On the right, a heatmap shows DNA methylation level (broadness and deepness for each kind of sequence context). The methylome data of human IMR90 fetal lung fibroblasts was used to represent the human.
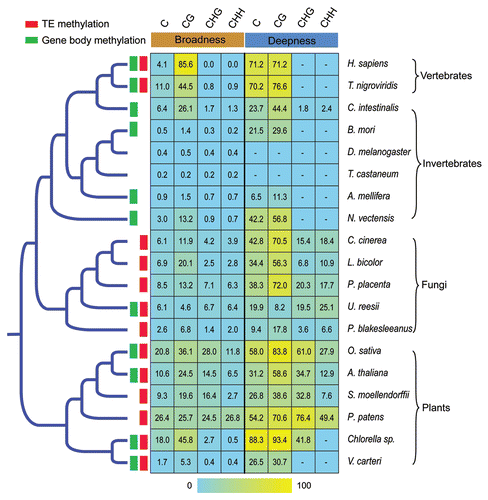
Table 1 Statistics and features of DNA methylation in vertebrates, invertebrates, plants and fungi
Table 2 Genome features and DNA methylation patterns of the available eukaryote methylomes