Abstract
The molecular requirements for neural tube closure are complex. This is illustrated by the occurrence of neural tube defects (NTDs) in many genetic mouse mutants, which implicate a variety of genes, pathways and cellular functions. NTDs are also prevalent birth defects in humans, affecting around 1 per 1000 pregnancies worldwide. In humans the causation is thought to involve the interplay of fetal genes and the effect of environmental factors. Recent studies on the aetiology of human NTDs, as well as analysis of mouse models, have raised the question of the possible involvement of epigenetic factors in determining susceptibility. A consideration of potential causative factors in human NTDs must now include both alterations in the regulation of gene expression, through mutation of promoter or regulatory elements, and the additional analysis of epigenetic regulation. Alterations in the epigenetic status can be directly modified by various environmental insults or maternal dietary factors.
Neural Tube Development and Disease
The neural tube is the embryonic precursor of the spinal cord and brain. It develops from a thickened region of the dorsal surface ectoderm, the neural plate, which undergoes a series of shaping and folding events. Briefly, the lateral edges of the neural plate elevate to form paired parallel folds running in an anterior to posterior orientation. Aided by bending along the body axis, these folds then converge and fuse along the dorsal midline to ultimately form the closed neural tube.Citation1,Citation2 Mammalian neural tube closure is a discontinuous process along the body axis, being initiated at discrete sites and progressing in a “zipper-like” fashion to close the open regions of neural folds, termed neuropores. The initial fusion site occurs at the hindbrain/cervical boundary, at around day 21 of human gestation (day 8 in mice), with closure spreading in both anterior and posterior directions. Another site of initiation occurs at the rostral limit of the neural plate, with progression in a caudal direction to meet the wave of closure from the hindbrain and seal the rostral neuropore, thereby completing closure in the brain.Citation3,Citation4 In mice, in which the closure process has been most extensively studied, there is a further initiation site at the midbrain-forebrain boundary such that two neuropores (anterior and hindbrain) are observed prior to final closure.Citation1,Citation5 Closure in the future spinal region proceeds caudally in a unidirectional manner, with final closure of the posterior neuropore at the mid-sacral level at day 26–28. At more caudal levels the neural tube is formed by a process known as secondary neurulation, where canalization of the tail bud region forms the sacro-caudal neural tube which becomes continuous with the primary neural tube.Citation2 The complete neural tube becomes populated by neurons and surrounded by the protective meninges and surrounding bone of the vertebrae.
Failure in initial fusion or progression of closure results in neural tube defects (NTDs), in which the neural ectoderm remains exposed and subject to degeneration. NTDs are among the most common of human birth defects, with an overall prevalence of around 0.5–2/1,000 pregnancies and frequently result in infant mortality or major health problems in surviving children.Citation6 The nature and severity of NTDs is determined by the stage and axial level at which closure fails.Citation2 Failure to initiate closure at the hindbrain/cervical boundary will result in the most severe defect, known as craniorachischisis, in which the neural folds remain splayed open throughout the midbrain, hindbrain and entire spinal region. Failure to complete closure of the rostral neuropore results in anencephaly, which may involve the midbrain alone, or also involve the hindbrain or more rarely the forebrain. Neither anencephaly nor craniorachischisis are compatible with life after birth, owing to in utero neurodegeneration. Failure of closure of the posterior neuropore leads to open spina bifida (myelomeningocele), a term which refers to the defects in the vertebral arches that obligatorily accompany the presence of the open neural folds. The clinical severity of spina bifida is affected by the axial level at which closure fails but patients frequently suffer paralysis of the legs, as well as bowel and bladder dysfunction.
Despite the high prevalence and traumatic consequences for affected individuals and their families, the underlying causes of NTD remain poorly understood in most individuals. In humans, identification of the precise biochemical and cellular factors involved has proved elusive, partly due to the lack of large families with Mendelian inheritance and also because of the high degree of heterogeneity between unrelated sporadic cases. It seems highly likely that the majority of NTD cases result from interaction between both genetic and environmental factors.Citation7–Citation9 Evidence for a genetic contribution is indicated by the high recurrence risk for siblings of affected individuals and association with specific chromosomal anomalies.Citation10–Citation12 The complexity of the molecular requirements for neural tube closure is well illustrated by the occurrence of NTDs in more than 200 different mouse genetic models.Citation13,Citation14 Moreover, the high penetrance observed on the original genetic background, is often seen to diminish if the mice are outcrossed indicating the contribution of modifier loci.Citation13 Only very few of the NTD-associated genes identified in mice have been confirmed to be involved in humans.Citation9 The majority of human NTD occur sporadically, with recurrences tending to fit a multifactorial polygenic or oligogenic pattern, rather than either dominant or recessive single gene inheritance with reduced penetrance.Citation13,Citation14 In addition to mutation of the coding sequence, altered transcriptional regulation of these genes has potential to cause NTDs. Indeed, in some cases, such as Grhl2, closure can be prevented by either loss of function or overexpression of a single gene.Citation15 A consideration of potential causative factors in human NTDs should therefore account for the possibility of deregulation of gene expression through epigenetic mechanisms.
A number of factors have been described to elevate the risk for NTDs. These include medical conditions such as maternal diabetesCitation16 or maternal obesity,Citation17 while environmental exposures such as cigarette smoke, mycotoxins or use of anti-epileptic drugs may have teratogenic effects.Citation18–Citation20 Maternal dietary factors leading to a high dietary glycemic index or a high glycemic load are associated with increased risk of an NTD affected pregnancy,Citation21 as are sub-optimal levels of folate and vitamin B12.Citation22,Citation23 Particular attention has been paid to the role of folate one-carbon metabolism, especially owing to the finding that maternal supplementation with folic acid reduces the risk of NTDs.Citation24–Citation26 Environmental factors may influence neural tube closure through a direct effect on embryonic metabolism/cell biology. Alternatively, some of the established risk factors may act mechanistically via an effect on epigenetic regulation thereby influencing susceptibility through altered gene expression. Evidence for a potential role for epigenetic effects on gene regulation is emerging both from the study of human NTDs and from analysis of mouse models.
Mechanisms of Epigenetic Regulation
The basis of epigenetic regulation of gene expression is complex, involving several interconnecting molecular layers referred to as the “epigenome.” These layers include DNA methylation, histone modifications involving post-translational covalent modifications such as acetylation, antisense RNAs, small interfering RNAs and also non-histone proteins that influence chromatin folding. All these processes regulate DNA transcription of specific genes but do not alter the primary sequence. The different mechanisms can act locally or with genome-wide effect, creating an epigenetic landscape reinforcing a transcriptionally favorable or unfavorable chromatin conformation.Citation27,Citation28 Some specific histone modifications can promote DNA methylation and vice versa.Citation29,Citation30 For example, methyl-CpG-binding protein MeCP2 attaches to methylated cytosines and attracts other proteins with enzymatic activity that promote nearby histone deacetylation.Citation31 The establishment and maintenance of epigenetic marks are not fully understood, but it is known that genetic variation in the DNA sequence itself, as well as the sequence of unlinked modifier loci, play additional roles.Citation32
DNA methylation in mammals occurs at C5 of the cytosine pyrimidine ring, within CpG dinucleotides, converting them to 5-methylcytosine. This covalent modification inhibits the affinity of methylation-sensitive DNA binding proteins, affects chromatin structure and usually correlates with transcriptional silencing.Citation33 In normal mammalian cells most of the genomic CpG sites (90–98%) are methylated, including exons, intergenic DNA and the mobile transposable elements called transposons.Citation34 Conversely, CpG-islands (CGIs), which are found in 50–60% of gene promoter regions, have a high density of CpG, are typically unmethylated and act as a regulation switch.Citation35 A gene can also be hypermethylated throughout but have a hypomethylated promoter region which controls its expression.Citation34
Epigenetic regulation has been found to operate very actively in normal mammal cell functioning, from conception to aging and death.Citation36 Reprogramming of DNA methylation of the totipotent zygote takes place in germ cells and during pre-implantationCitation35 (). Mammal sperm and egg genomes are highly methylated when compared with somatic cells. However, a few hours after fertilization, rapid demethylation of the paternal genome takes place by active but yet undefined mechanisms, in addition to histone modification acquisition.Citation37 The maternal genome follows a slower and more passive process, with demethylation by simple dilution of DNA methylation during replication, preventing DNMT activity at the replication fork.Citation38 Up to the morula stage, DNA methylation remains reduced and cells are pluripotent, with all genes potentially active. Simultaneously, primordial germ cells (PGCs) undergo changes in histones and reorganization of chromatin. After implantation, genome-wide resetting occurs for most of the genome in a lineage-specific manner and continues, to a lesser extent over the rest of the fetal development (). Re-methylation at this stage varies upon the part of the embryo concerned and, whereas ectoderm and mesoderm become hypermethylated, primary endoderm and trophoblast remain hypomethylated. There seems to be a sequence of re-methylation dictating the structure and function of each formatting somatic tissue.Citation39
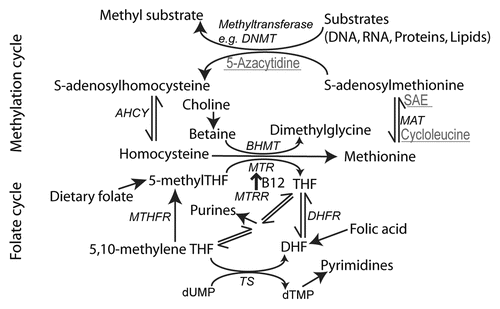
The methyl groups used for DNA methylation are supplied by S-adenosylmethionine (SAM), and are catalyzed by various types of DNA methyltransferases (DNMTs) (). De novo methylation patterns during gametogenesis, embryogenesis and tissue differentiation are established mainly by the enzymes DNMT3A and DNMT3B, whose activity is downregulated upon differentiation of embryonic stem cells and remain low during adult life. Maintenance of these patterns in somatic cells is dependent on different DNMT1 variants, which also have some de novo activity.Citation40 The accuracy of copying DNA methylation patterns at cell division is estimated at ∼96%. DNMT enzymes are believed to have a dual role, both in methylation and demethylation.Citation41,Citation42 Recent studies have also suggested that demethylation of DNA is indirect with modification of the methyl-cytosine by deamination or oxidation, sometimes referred to as ‘hydroxymethlyation’, followed by DNA repair.Citation43
Several lines of evidence from epidemiologic studies are suggestive of a link between impaired methylation cycle and human NTDs. These include the association with elevated homocysteine and sub-optimal levels of folate and/or vitamin B12 in maternal blood.Citation23,Citation44–Citation46 Probably the most extensively studied genetic risk factor for NTDs is the 677C>T SNP in 5, 10-methylenetetrahydrofolate reductase (MTHFR) (reviewed in ref. Citation47), which is associated with elevated homocysteine,Citation48 presumably due to diminished production of 5-methyl THF (see below). An alternative folate-independent mechanism for re-methylation of homocysteine involves transfer of a one-carbon unit from betaine, catalyzed by betaine homocysteinemethyltransferase (BHMT) (). It is intriguing that higher maternal dietary intake of betaine and choline, the precursor of betaine, shows a striking association with reduced risk of NTDs.Citation49 Furthermore, choline levels were found to be correlated with NTD risk in analysis of second trimester maternal serum, where low levels increases risk and high levels are protective.Citation50 Interestingly, the study population was North American and therefore the diet was folate-fortified, such that other one-carbon metabolites including homocysteine, methionine, folate, vitamin B12 and betaine were not found to be altered in NTD pregnancies. Inhibition of choline uptake or metabolism causes NTDs in cultured mouse embryos.Citation51 Whether these defects relate to a methylation deficit or lack of phosphatidylcholine synthesis is not entirely clear. The lack of association between betaine level and risk of human NTDs perhaps argues against a methylation-related mechanism, but further analysis of the relationship between maternal choline/betaine and embryonic levels would be merited. Later in development, dietary imposition of choline deficiency has been found to alter DNA and histone methylation patterns in fetal mouse hippocampus.Citation52,Citation53
Evidence that Folate One-Carbon Metabolism Influences Risk of NTDs via its Role in Methylation?
Several lines of evidence suggest that folate one-carbon metabolism is a key determinant of NTD susceptibility (reviewed by ref. Citation47 and Citation54–Citation56). However, NTDs do not appear to simply result from a maternal folate deficiency (which is corrected by folic acid supplementation), since maternal levels in most affected pregnancies fall within the “normal” range.Citation57 Moreover, in experimental models of folate deficiency, the developing mouse embryo appears relatively resistant to maternal folate depletion. Whereas profound dietary folate deficiency affects embryonic folate levels at neurulation stages, a moderate deficiency produces significant effects on maternal circulating folate and homocysteine but has no apparent effect on embryonic folate content.Citation58 Therefore, it appears likely that supplemental folic acid acts either to overcome a defect in embryonic folate one-carbon metabolism or to compensate for a predisposing factor that itself may be unrelated to folate status.
Circulating folate, principally in the form of 5-methyl tetrahydrofolate (5-MeTHF), is taken up into cells by folate receptors (FOLR1 and FOLR2) or by the reduced folate carrier (RFC1). Mice that are deficient for Folr1 develop NTDs but can be rescued from early lethality by folic acid treatment, thereby demonstrating that folate uptake is essential for embryonic development.Citation59,Citation60 Within the cell, folates act as cofactors in a network of reactions for the transfer of one-carbon units, termed “folate one-carbon metabolism,” that is essential for the production of purines and pyrimidines ().Citation55 Defective thymidylate biosynthesis has been implicated in NTDs in humans and mouse models,Citation61–Citation63 suggesting that adequate supply of nucleotides for cellular proliferation is essential for closure of the neural tube.
In addition to nucleotide biosynthesis, folate one-carbon metabolism also generates SAM, which as mentioned above is the methyl donor for methylation of DNA, proteins, RNA and lipids and hence a key requirement for epigenetic regulatory mechanisms. It is therefore not surprising that research on NTDs has also focused on this aspect as a key function of the folate cycle. It is thought that suboptimal folate one-carbon metabolism could be associated with an increased risk of NTDs, directly as a result of diminished or decreased essential methylation.Citation64,Citation65 The folate and methylation cycles are interlinked by the transfer of a one-carbon unit from 5-methyl tetrahydrofolate to homocysteine catalyzed by methionine synthase, for which vitamin B12 (cobalamin) acts as cofactor. This reaction serves to generate methionine and tetrahydrofolate (THF) and is important for methylation but also for folate cycle flux, as the reaction that generates 5-methyl THF is essentially irreversible (). Inhibition of methionine synthase would result in accumulation of homocysteine, but also 5-methyl THF and therefore effective depletion of other folates, the so-called methyl trap.Citation47,Citation55
Are the Predicted Correlations Between Biomarkers of One-Carbon Metabolism and Methylation Born Out by Analysis of DNA in Humans?
Evidence for a potential link between one-carbon metabolites and the fetal epigenome has been suggested by small scale studies in which DNA methylation and one-carbon metabolites were assayed in cord blood, collected at term following normal pregnancy.Citation66,Citation67 Methylation of long interspersed nucleotide element-1 (LINE-1) sequences, assayed as an indicator of genome wide DNA methylation, was not associated with either use of folic acid supplements or serum folate. However, there was a significant inverse correlation with fetal plasma homocysteine level and this correlation was association was also apparent in more detailed analysis of CpG methylation patterns.Citation66,Citation67 While few neurulation-stage human embryos are available for study, initial data on DNA methylation in NTDs is now emerging from analysis of fetal DNA of nervous tissue (or its remnants) following termination of pregnancy.Citation68 Methylation of genomic DNA (5-methyl cytosine content) and LINE-1 sequences was found to be lower among NTD cases involving the cranial region compared to controls, but not among spina bifida cases.Citation68 The reduction in methylation was associated with reduced maternal vitamin B12 level, but not folate status, although a subsequent study shows an inverse correlation between methylation of genomic DNA in abortus brain tissue and maternal serum folate.Citation69
Does the Experimental Evidence from Model Systems Support the Hypothesis that Diminished Methylation Could Contribute to the Development of NTDs?
Cranial NTDs arise in homozygous null embryos for Dnmt3bCitation70 and in embryos cultured in the presence of 5-azacytidine,Citation71 suggesting that there is a requirement for DNA methylation in neural tube closure. Similarly, exposure to methylation cycle inhibitors or excess methionine causes cranial NTDs without other major defects, even in non-mutant strains.Citation72,Citation73 Both these treatments result in a reduced ratio of S-adenosylmethionine (SAM) to S-adenosylhomocysteine (SAH), indicative of reduced methylation potential. A corresponding reduction in global DNA methylation was observed in embryos treated with the methylation cycle inhibitor, ethionine.Citation58 On the other hand, NTDs are not observed in null embryos for Mthfr, despite a significant reduction in SAM/SAH ratio and global DNA methylation.Citation74 Therefore, evidence that the apparent association of MTHFR polymorphism with human NTDs is mediated through an epigenetic mechanism is currently unsupported. Similarly, NTDs are not observed in mice carrying a hypomorphic allele of Mtrr, encoding methionine synthase reductase, which is required for methionine synthase activity,Citation75 although there is some evidence of an association with NTDs in humans.Citation9,Citation76 These models emphasize the point that elevated homocysteine is likely to be a marker of impaired one carbon metabolism in NTDs rather than directly causative. This is further supported by the fact that homocysteine treatment of mouse embryos developing in culture or in vivo does not cause NTDs.Citation77,Citation78
Folic acid treatment can alter global DNA methylation in adult rats.Citation79 It is also apparent that genomic DNA methylation can be altered in the offspring of mice fed a methyl donor-rich diet, at least at transposable elements.Citation80 However, the converse situation of folate deficiency is less clear. NTDs do not arise in wild-type mouse embryos under conditions of profound maternal folate-deficiency, which is imposed by both folate-deficient diet and treatment with antibiotics (that removes gut flora that synthesize folates), despite a significant reduction in SAM/SAH ratio.Citation58,Citation81 Folate-deficiency can, however, exacerbate susceptibility in embryos that carry a genetic predisposition to NTDs, such as splotch (Pax3) mutants. In this model there was no apparent effect of reduced SAM/SAH ratio on global DNA methylation, although an effect on specific loci cannot be ruled out.Citation58 Similarly, overall reduction in methylation does not increase the frequency of NTDs in splotch embryos made doubly mutant for Mthfr null alleles.Citation82 Maternal folate deficiency also induces cranial NTDs in mouse embryos with a curly tail genetic background.Citation83 Again, a mechanism in which overall impairment of DNA methylation causes NTDs appears unlikely, since breeding the Mthfr null allele into the curly tail strain does not increase the frequency of defects despite major reduction in SAM/SAH ratio.Citation84
Histone Methylation and NTDs
To date few studies have addressed the possible role of altered histone methylation in development of NTDs. However, a recent study demonstrated increased methylation of lysine 27 of histone H3 (H3K27) in cultured neural crest cells and neural tube explants from splotch (Pax3) mutant embryos, which develop NTDs.Citation85 Interestingly, this abnormality was not observed following treatment with folic acid, that is known to rescue splotch NTDs,Citation62 raising the possibility that splotch NTDs could be associated with altered epigenetic regulation. The role of folic acid in this context appears complex since increased folic acid has previously been associated with elevated DNA methylation potential. The finding of diminished H3K27 methylation following folic acid treatment therefore appears likely to reflect prevention of the underlying defect rather than a direct effect via stimulation of methylation.
Histone Acetylation and NTDs
In addition to methylation, other histone modifications can also affect chromatin function and thereby contribute to modulation of gene expression.Citation86 In the context of neural tube closure, regulation of acetylation appears to play a critical role. In addition to N-terminal acetylation, which occurs cotranslationally on most proteins, some proteins are subject to post-translational acetylation of specific lysine residues. Acetylation neutralizes the positive charge on the lysine amino group and can therefore influence electrostatic properties and function of the protein. The acetylation status of specific proteins depends on an equilibrium between the activity of histone acetylases (HATs) and deacetylases (HDACs), which respectively add and remove acetyl groups.Citation86,Citation87 Whereas HATs (such as p300) frequently act as transcriptional co-activators, HDAC activity results in chromatin compaction and transcriptional repression.Citation88
The observation of cranial NTDs among knock-out embryos for p300 suggested that histone acetylase activity is essential for neural tube closure.Citation87 Further evidence of a requirement for acetylation in neural tube closure was provided by generation of a knock-in allele of the acetyltransferase encoding Kat2a (Gcn5) gene. The Gcn5hat allele carries point mutations in the catalytic domain that abolish HAT activity.Citation89 In contrast to embryos that are completely null for Gcn5, which die early in gestation, Gcn5hat/hat embryos survive beyond neurulation stages but exhibit cranial NTDs,Citation89 as do mice carrying a hypomorphic allele of Gcn5 with reduced expression.Citation90 Cranial NTDs also occur in knockout embryos for Cited2, encoding a member of the CITED (CBP/p300 interacting transactivator with ED-rich tail) protein family, which, as its name suggests, can bind to p300 and its paralogue CBP (cAMP-responsive element-binding protein).Citation91,Citation92 These interactions may interfere with, and thereby modulate the acetylation of, p300 target proteins such as HIF-1α. Interestingly, HIF-1α-responsive genes are upregulated in Cited2 null embryos.Citation92,Citation93
The cellular and developmental mechanisms underlying NTDs in acetylation-related mouse models are not completely clear. A proportion of Gcn5hat/hat embryos are growth retarded and exhibit increased rates of apoptosis. However, NTDs still arise even in those embryos in which overall growth, neuroepithelial proliferation and apoptosis are comparable to wild-types.Citation89 NTDs in Cited2 mutants are associated with a dramatic increase in apoptosis in the neural folds prior to failure of closure and could therefore potentially play a causative role.Citation94 Nevertheless, prevention of NTDs by folic acid in Cited2 mutants is not associated with an obvious reduction in apoptosis, suggesting either that apoptosis is not causative or that folic acid does not act by amelioration of the underlying defect.Citation94
Changes in regulation of acetylation has also been implicated in failure of neural tube closure owing to the teratogenic effect of pharmacological inhibitors of HDACs, such as valproic acid (VPA) and trichostatin A, which cause NTDs as well as defects of the axial skeleton.Citation95,Citation96 In accordance with a downstream effect mediated through HDAC inhibition, expression analyses of valproic acid-treated embryos showed misregulation of the HDAC ontology group and elevated acetylation of histone H4 in the neural tube.Citation96–Citation98 Among VPA derivatives, HDAC activity correlates with teratogenic potential.Citation99 Knock-out of Hdac1 results in early embryonic lethality with reduced proliferation, probably due to upregulation of p21.Citation100 Altered proliferation would have potential to have an impact on neural tube closure. However, while conditional mutants of both Hdac1 and Hdac2 have been generated and show a requirement in neuronal development,Citation101 we are not aware that experiments to selectively ablate their activity in the neural folds have yet been performed. However, a proportion of Hdac4 mutants are reported to develop cranial NTDs,Citation102 as do some knock-out embryos for Sirt1, another histone deacetylase.Citation103
Overall, it appears that both decreased acetylation (HAT mutants) and increased acetylation (HDAC mutant or inhibitor-treated) are associated with development of NTDs. When interpreting the mechanisms by which acetylation influences neural tube closure it should be considered that, while many of the functions of HATs are mediated through histones, it is also apparent that other proteins are also targets for acetylation/deacetylation.Citation104,Citation105 In the context of neural tube closure, proteins such as p53 and Rb are of interest owing to their role in regulation of cell cycle progression and apoptosis.Citation103,Citation104 Acetylation may also mediate interplay between different mechanisms of epigenetic regulation. For example, studies in yeast indicate that the SWI/SNF chromatin remodeling complex is regulated by acetylation of its constituent proteins in addition to histone acetylation.Citation106
Chromatin Remodeling and NTDs
Although not directly participating in chromatin modifications, three types of proteins are critical for epigenetic regulation: chromatin remodeling complexes, effector binding-proteins and insulator proteins.Citation35 Specific CpG-binding proteins such as MeCP2 interpret DNA methylation by recruiting, transcribing or repressing complexes that decipher the histone/DNA methylation marks. Polycomb-group proteins can remodel chromatin and target human gene promoters, throughout development.Citation107 Other factors which are also involved in chromatin structure and gene regulation include nucleosome positioning, especially in the vicinity of the transcription start site results in inactivation or activation respectively, maybe due to affecting access for binding of transcription factors.Citation108
The occurrence of NTDs in several mouse mutants for chromatin remodeling enzymesCitation13,Citation14 further emphasizes the multiplicity of mechanisms by which transcriptional regulation may be altered with potential detrimental effects on closure. Chromatin binding/remodeling proteins associated with mouse NTDs include SMARCA4 (BRG1);Citation109 CERCR2;Citation110 SMARCC1 (SRG3).Citation111 Cranial NTDs also occur at high frequency, in association with profound growth retardation, in embryos lacking BRD2 (bromodomain-containing protein 2),Citation112 which binds acetylated histones and chromatin remodeling proteins. Composition of the chromatin remodeling complex provides differential specificity of binding to target loci.Citation113,Citation114 Interrogation of data-sets generated from recent genome-wide mapping screens for targets and interactors of chromatin remodeling complexes may therefore inform further analysis of the mechanism by which loss of function of some components (e.g., BRG1) results in NTDs.
Summary
Studies in model organisms have provided valuable insight into the developmental and cellular basis of neural tube closure. Similarities in the anatomy and pathogenesis of mouse and human NTDs imply that there is considerable overlap in the underlying causative mechanisms. However, despite NTDs being common birth defects in humans, in most cases the underlying molecular pathology continues to remain obscure. This is perhaps surprising given the large number of mouse single gene mutants that develop NTDs with high penetrance. The most likely explanation lies in the apparent multigenic inheritance of human NTDs, under the influence of modifier genes and environmental factors. In addition to the large number of potential candidate genes, further complexity comes from the fact that in addition to coding mutations there may be causative involvement of gene expression, perhaps resulting from regulatory sequence variants or altered epigenetic architecture. Among the many different epigenetic mechanisms known to be involved in early development, the best understood is currently that of DNA methylation. Animal studies suggest that impaired DNA methylation can interfere with normal neural tube closure and preliminary data indicate that it may potentially contribute to human NTDs. Other key mechanisms such as histone modifications and chromatin remodeling have also been associated with NTDs in mice and, at least in the case of histone acetylation, in humans as well. It is well known that phenotypic expression of the developing embryo is strongly influenced by the maternal environment, especially diet. This raises the possibility that the relationship between folic acid supplementation and/or folate one-carbon metabolism with the risk of NTDs may be mediated in part through their effects on methylation. In this respect, there is certainly experimental evidence to show that DNA methylation can be increased following folic acid supplementation, but it is less clear whether sub-optimal folate status results in diminished methylation. The next challenge will be to determine whether maternal dietary factors that affect NTD risk do so by altering epigenetic regulation, and then to identify the key genes and pathways that are differentially regulated in this way.
Figures and Tables
Figure 1 Methylation changes during early development. The epigenome is a dynamic process where rapid de-methylation occurs immediately post fertilization but is then followed by re-methylation in the blastocyst and early embryo. Developmental stages are shown diagrammatically. During neurulation stages, the direction of neural tube fusion is shown by arrows, resulting in closure of the rostral neuropore (dark shaded area) in the cranial region and the posterior neuropore in the caudal region. Craniorachischisis results from failure to initiate closure of the neural folds, anencephaly and spina bifida from incomplete closure in cranial and caudal regions respectively.
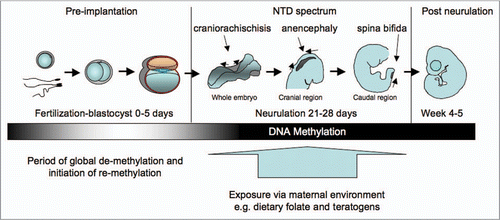
Figure 2 Summary of folate one-carbon metabolism. A simplified diagram showing the key functions of the folate cycle (lower half of diagram), involving transfer of 1C groups between folate molecules, required for pyrimidine and purine biosynthesis. The action of MTHFR (5,10-methylene tetrahydrofolate reductase) produces 5-methylTHF for re-methylation of homocysteine by MTR (methionine synthase). Alternatively, homocysteine is re-methylated by the action of BHMT (betaine-homocysteine methyltransferase). In the methylation cycle (upper half), S-adenosylmethionine (SAM) acts as the methyl group donor in a variety of methylation reactions. Reagents that inhibit steps of the methylation cycle and produce NTDs in cultured mouse embryos are shown in grey and underlined. S-adenosylethionine (SAE) is a non-metabolized analogue of SAM. Enzymes are shown in italics. AHCY, S-adenosylhomocysteine hydrolase; DHFR, dihydrofolate reductase; MAT, methionine adenosyltransferase; MTRR, methionine synthase reductase; TS, thymidylate synthase.
Acknowledgments
The authors would like to thank the Wellcome Trust, Medical Research Council, SPARKS, WellBeing of Woman and the UCL Biomedical Research Centre for funding research in their laboratories. P.S. is supported by Great Ormond Street Hospital Children's Charity.
References
- Copp AJ, Greene NDE, Murdoch JN. The genetic basis of mammalian neurulation. Nat Rev Genet 2003; 4:784 - 793
- Greene ND, Copp AJ. Development of the vertebrate central nervous system: formation of the neural tube. Prenatal Diag 2009; 29:303 - 311
- Moore CA. Wyszynski DF. Classification of neural tube defects. Neural Tube Defects: From Origin to Treatment 2006; Oxford Oxford University Press 66 - 75
- Copp AJ, Greene NDE. Genetics and development of neural tube defects. J Pathol 2010; 220:217 - 230
- Copp AJ. Neurulation in the cranial region—normal and abnormal. J Anat 2005; 207:623 - 635
- Mitchell LE. Epidemiology of neural tube defects. Am J Med Genet C Semin Med Genet 2005; 135:88 - 94
- Holmes LB, Driscoll SG, Atkins L. Etiologic heterogeneity of neural-tube defects. N Engl J Med 1976; 294:365 - 369
- Detrait ER, George TM, Etchevers HC, Gilbert JR, Vekemans M, Speer MC. Human neural tube defects: developmental biology, epidemiology and genetics. Neurotoxicol Teratol 2005; 27:515 - 524
- Greene NDE, Stanier P, Copp AJ. Genetics of human neural tube defects. Hum Mol Gene 2009; 18:113 - 129
- Kennedy D, Chitayat D, Winsor EJT, Silver M, Toi A. Prenatally diagnosed neural tube defects: Ultrasound, chromosome and autopsy or postnatal findings in 212 cases. Am J Med Genet 1998; 77:317 - 321
- Chen CP. Chromosomal abnormalities associated with neural tube defects (I): full aneuploidy. Taiwan J Obstet Gynecol 2007; 46:325 - 335
- Chen CP. Chromosomal abnormalities associated with neural tube defects (II): partial aneuploidy. Taiwan J Obstet Gynecol 2007; 46:336 - 351
- Harris MJ, Juriloff DM. An update to the list of mouse mutants with neural tube closure defects and advances toward a complete genetic perspective of neural tube closure. Birth Defects Res A Clin Mol Teratol 2010; 88:653 - 669
- Harris MJ, Juriloff DM. Mouse mutants with neural tube closure defects and their role in understanding human neural tube defects. Birth Defects Res A Clin Mol Teratol 2007; 79:187 - 210
- Brouns MR, De Castro SC, Terwindt-Rouwenhorst EA, Massa V, Hekking JW, Hirst CS, et al. Overexpression of Grhl2 causes spina bifida in the Axial defects mutant mouse. Hum Mol Genet 2011; 20:1536 - 1546
- Eriksson UJ, Cederberg J, Wentzel P. Congenital malformations in offspring of diabetic mothers—animal and human studies. Rev Endocr Metab Disord 2003; 4:79 - 93
- Hendricks KA, Nuno OM, Suarez L, Larsen R. Effects of hyperinsulinemia and obesity on risk of neural tube defects among Mexican Americans. Epidemiology 2001; 12:630 - 635
- Suarez L, Ramadhani T, Felkner M, Canfield MA, Brender JD, Romitti PA, et al. Maternal smoking, passive tobacco smoke and neural tube defects. Birth Defects Res A Clin Mol Teratol 2011; 91:29 - 33
- Marasas WF, Riley RT, Hendricks KA, Stevens VL, Sadler TW, Gelineau-van Waes J, et al. Fumonisins disrupt sphingolipid metabolism, folate transport and neural tube development in embryo culture and in vivo: a potential risk factor for human neural tube defects among populations consuming fumonisin-contaminated maize. J Nutr 2004; 134:711 - 716
- Buehler BA, Rao V, Finnell RH. Biochemical and molecular teratology of fetal hydantoin syndrome. Neurol Clin 1994; 12:741 - 748
- Yazdy MM, Liu S, Mitchell AA, Werler MM. Maternal dietary glycemic intake and the risk of neural tube defects. Am J Epidemiol 2009; 171:407 - 414
- Smithells RW, Sheppard S, Schorah CJ. Vitamin deficiencies and neural tube defects. Arch Dis Child 1976; 51:944 - 950
- Kirke PN, Molloy AM, Daly LE, Burke H, Weir DG, Scott JM. Maternal plasma folate and vitamin B12 are independent risk factors for neural tube defects. Q J Med 1993; 86:703 - 708
- Smithells RW, Sheppard S, Schorah CJ, Seller MJ, Nevin NC, Harris R, et al. Possible prevention of neural-tube defects by periconceptional vitamin supplementation. Lancet 1980; 1:339 - 340
- Wald N, Sneddon J, Densem J, Frost C, Stone R. MRC Vitamin Study Res Group. Prevention of neural tube defects: Results of the Medical Research Council Vitamin Study. Lancet 1991; 338:131 - 137
- Czeizel AE, Dudás I. Prevention of the first occurrence of neural-tube defects by periconceptional vitamin supplementation. N Engl J Med 1992; 327:1832 - 1835
- Waterland RA, Michels KB. Epigenetic epidemiology of the developmental origins hypothesis. Annu Rev Nutr 2007; 27:363 - 388
- Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis 2010; 31:27 - 36
- Burdge GC, Lillycrop KA. Nutrition, epigenetics and developmental plasticity: implications for understanding human disease. Annu Rev Nutr 2010; 30:315 - 339
- Vaissiere T, Sawan C, Herceg Z. Epigenetic interplay between histone modifications and DNA methylation in gene silencing. Mutat Res 2008; 659:40 - 48
- Clouaire T, Stancheva I. Methyl-CpG binding proteins: specialized transcriptional repressors or structural components of chromatin?. Cell Mol Life Sci 2008; 65:1509 - 1522
- Whitelaw NC, Whitelaw E. How lifetimes shape epigenotype within and across generations. Hum Mol Genet 2006; 15:131 - 137
- Guil S, Esteller M. DNA methylomes, histone codes and miRNAs: tying it all together. Int J Biochem Cell Biol 2009; 41:87 - 95
- Zeisel SH. Epigenetic mechanisms for nutrition determinants of later health outcomes. Am J Clin Nutr 2009; 89:1488 - 1493
- Kim JK, Samaranayake M, Pradhan S. Epigenetic mechanisms in mammals. Cell Mol Life Sci 2009; 66:596 - 612
- Delcuve GP, Rastegar M, Davie JR. Epigenetic control. J Cell Physiol 2009; 219:243 - 250
- Morgan HD, Santos F, Green K, Dean W, Reik W. Epigenetic reprogramming in mammals. Hum Mol Genet 2005; 14:47 - 58
- Kelly TL, Trasler JM. Reproductive epigenetics. Clin Genet 2004; 65:247 - 260
- Junien C, Nathanielsz P. Report on the IASO Stock Conference 2006: early and lifelong environmental epigenomic programming of metabolic syndrome, obesity and type II diabetes. Obes Rev 2007; 8:487 - 502
- Jones PA, Liang G. Rethinking how DNA methylation patterns are maintained. Nat Rev Genet 2009; 10:805 - 811
- Bird A. Perceptions of epigenetics. Nature 2007; 447:396 - 398
- Métivier R, Gallais R, Tiffoche C, Le Péron C, Jurkowska RZ, Carmouche RP, et al. Cyclical DNA methylation of a transcriptionally active promoter. Nature 2008; 452:45 - 50
- Popp C, Dean W, Feng S, Cokus SJ, Andrews S, Pellegrini M, et al. Genome-wide erasure of DNA methylation in mouse primordial germ cells is affected by AID deficiency. Nature 2010; 463:1101 - 1105
- Steegers-Theunissen RP, Boers GH, Trijbels FJ, Finkelstein JD, Blom HJ, Thomas CM, et al. Maternal hyperhomocysteinemia: A risk factor for neural tube defects. Metabolism 1994; 43:1475 - 1480
- Mills JL, McPartlin JM, Kirke PN, Lee YJ, Conley MR, Weir DG, et al. Homocysteine metabolism in pregnancies complicated by neural-tube defects. Lancet 1995; 345:149 - 151
- Ray JG, Wyatt PR, Thompson MD, Vermeulen MJ, Meier C, Wong PY, et al. Vitamin B12 and the risk of neural tube defects in a folic-acid-fortified population. Epidemiology 2007; 18:362 - 366
- Molloy AM, Brody LC, Mills JL, Scott JM, Kirke PN. The search for genetic polymorphisms in the homocysteine/folate pathway that contribute to the etiology of human neural tube defects. Birth Defects Res A Clin Mol Teratol 2009; 85:285 - 294
- van der Put NM, Steegers-Theunissen RP, Frosst P, Trijbels FJ, Eskes TK, van den Heuvel LP, et al. Mutated methylenetetrahydrofolate reductase as a risk factor for spina bifida. Lancet 1995; 346:1070 - 1071
- Shaw GM, Carmichael SL, Yang W, Selvin S, Schaffer DM. Periconceptional dietary intake of choline and betaine and neural tube defects in offspring. Am J Epidemiol 2004; 160:102 - 109
- Shaw GM, Finnell RH, Blom HJ, Carmichael SL, Vollset SE, Yang W, et al. Choline and risk of neural tube defects in a folate-fortified population. Epidemiology 2009; 20:714 - 719
- Fisher MC, Zeisel SH, Mar MH, Sadler TW. Perturbations in choline metabolism cause neural tube defects in mouse embryos in vitro. FASEB J 2002; 16:619 - 621
- Niculescu MD, Craciunescu CN, Zeisel SH. Dietary choline deficiency alters global and gene-specific DNA methylation in the developing hippocampus of mouse fetal brains. FASEB J 2006; 20:43 - 49
- Mehedint MG, Niculescu MD, Craciunescu CN, Zeisel SH. Choline deficiency alters global histone methylation and epigenetic marking at the Re1 site of the calbindin 1 gene. FASEB J 2010; 24:184 - 195
- Blom HJ, Shaw GM, Den Heijer M, Finnell RH. Neural tube defects and folate: case far from closed. Nat Rev Neurosci 2006; 7:724 - 731
- Beaudin AE, Stover PJ. Insights into metabolic mechanisms underlying folate-responsive neural tube defects: A minireview. Birth Defects Res A Clin Mol Teratol 2009; 85:274 - 284
- Greene ND, Massa V, Copp AJ. Understanding the causes and prevention of neural tube defects: Insights from the splotch mouse model. Birth Defects Res A Clin Mol Teratol 2009; 85:322 - 330
- Scott JM. Folate and vitamin B12. Proc Nutr Soc 1999; 58:441 - 448
- Burren KA, Savery D, Massa V, Kok RM, Scott JM, Blom HJ, et al. Gene-environment interactions in the causation of neural tube defects: folate deficiency increases susceptibility conferred by loss of Pax3 function. Hum Mol Genet 2008; 17:3675 - 3685
- Piedrahita JA, Oetama B, Bennett GD, van Waes J, Kamen BA, Richardson J, et al. Mice lacking the folic acid-binding protein Folbp1 are defective in early embryonic development. Nature Genet 1999; 23:228 - 232
- Spiegelstein O, Mitchell LE, Merriweather MY, Wicker NJ, Zhang Q, Lammer EJ, et al. Embryonic development of folate binding protein-1 (Folbp1) knockout mice: Effects of the chemical form, dose and timing of maternal folate supplementation. Dev Dyn 2004; 231:221 - 231
- Dunlevy LP, Chitty LS, Burren KA, Doudney K, Stojilkovic-Mikic T, Stanier P, et al. Abnormal folate metabolism in foetuses affected by neural tube defects. Brain 2007; 130:1043 - 1049
- Fleming A, Copp AJ. Embryonic folate metabolism and mouse neural tube defects. Science 1998; 280:2107 - 2109
- Beaudin AE, Abarinov EV, Noden DM, Perry CA, Chu S, Stabler SP, et al. Shmt1 and de novo thymidylate biosynthesis underlie folate-responsive neural tube defects in mice. Am J Clin Nutr 2011; 93:789 - 798
- Scott JM, Weir DG, Molloy A, McPartlin J, Daly L, Kirke P. Bock G, Marsh J. Folic acid metabolism and mechanisms of neural tube defects. Neural Tube Defects (Ciba Foundation Symposium 181) 1994; Chichester John Wiley & Sons 180 - 187
- Finnell RH, Blom HJ, Shaw GM. Does global hypomethylation contribute to susceptibility to neural tube defects?. Am J Clin Nutr 2010; 91:1153 - 1154
- Fryer AA, Nafee TM, Ismail KM, Carroll WD, Emes RD, Farrell WE. LINE-1 DNA methylation is inversely correlated with cord plasma homocysteine in man: a preliminary study. Epigenetics 2009; 4:394 - 398
- Fryer AA, Emes RD, Ismail KM, Haworth KE, Mein C, Carroll WD, et al. Quantitative, high-resolution epigenetic profiling of CpG loci identifies associations with cord blood plasma homocysteine and birth weight in humans. Epigenetics 2011; 6:86 - 94
- Wang L, Wang F, Guan J, Le J, Wu L, Zou J, et al. Relation between hypomethylation of long interspersed nucleotide elements and risk of neural tube defects. Am J Clin Nutr 2010; 91:1359 - 1367
- Chang H, Zhang T, Zhang Z, Bao R, Fu C, Wang Z, et al. Tissue-specific distribution of aberrant DNA methylation associated with maternal low-folate status in human neural tube defects. J Nutr Biochem 2011; In press
- Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999; 99:247 - 257
- Matsuda M. Comparison of the incidence of 5-azacytidine-induced exencephaly between MT/HokIdr and Slc:ICR mice. Teratology 1990; 41:147 - 154
- Dunlevy LPE, Burren KA, Chitty LS, Copp AJ, Greene NDE. Excess methionine suppresses the methylation cycle and inhibits neural tube closure in mouse embryos. FEBS Lett 2006; 580:2803 - 2807
- Dunlevy LPE, Burren KA, Mills K, Chitty LS, Copp AJ, Greene NDE. Integrity of the methylation cycle is essential for mammalian neural tube closure. Birth Defects Res A 2006; 76:544 - 552
- Chen Z, Karaplis AC, Ackerman SL, Pogribny IP, Melnyk S, Lussier-Cacan S, et al. Mice deficient in methylenetetrahydrofolate reductase exhibit hyperhomocysteinemia and decreased methylation capacity, with neuropathology and aortic lipid deposition. Hum Mol Genet 2001; 10:433 - 443
- Elmore CL, Wu X, Leclerc D, Watson ED, Bottiglieri T, Krupenko NI, et al. Metabolic derangement of methionine and folate metabolism in mice deficient in methionine synthase reductase. Mol Genet Metab 2007; 91:85 - 97
- van der Linden IJ, den Heijer M, Afman LA, Gellekink H, Vermeulen SH, Kluijtmans LA, et al. The methionine synthase reductase 66A>G polymorphism is a maternal risk factor for spina bifida. J Mol Med 2006; 84:1047 - 1054
- Greene NDE, Dunlevy LE, Copp AJ. Homocysteine is embryotoxic but does not cause neural tube defects in mouse embryos. Anat Embryol 2003; 206:185 - 191
- Bennett GD, Vanwaes J, Moser K, Chaudoin T, Starr L, Rosenquist TH. Failure of homocysteine to induce neural tube defects in a mouse model. Birth Defects Res B Dev Reprod Toxicol 2006; 77:89 - 94
- Iskandar BJ, Rizk E, Meier B, Hariharan N, Bottiglieri T, Finnell RH, et al. Folate regulation of axonal regeneration in the rodent central nervous system through DNA methylation. J Clin Invest 2010; 120:1603 - 1616
- Waterland RA, Jirtle RL. Transposable elements: targets for early nutritional effects on epigenetic gene regulation. Mol Cell Biol 2003; 23:5293 - 5300
- Burgoon JM, Selhub J, Nadeau M, Sadler TW. Investigation of the effects of folate deficiency on embryonic development through the establishment of a folate deficient mouse model. Teratology 2002; 65:219 - 227
- Pickell L, Li D, Brown K, Mikael LG, Wang XL, Wu Q, et al. Methylenetetrahydrofolate reductase deficiency and low dietary folate increase embryonic delay and placental abnormalities in mice. Birth Defects Res A Clin Mol Teratol 2009; 85:531 - 541
- Burren KA, Scott JM, Copp AJ, Greene ND. The genetic background of the curly tail strain confers susceptibility to folate-deficiency-induced exencephaly. Birth Defects Res A Clin Mol Teratol 2010; 88:76 - 83
- De Castro SC, Leung KY, Savery D, Burren K, Rozen R, Copp AJ, et al. Neural tube defects induced by folate deficiency in mutant curly tail (Grhl3) embryos are associated with alteration in folate one-carbon metabolism but are unlikely to result from diminished methylation. Birth Defects Res A Clin Mol Teratol 2010; 88:612 - 618
- Ichi S, Costa FF, Bischof JM, Nakazaki H, Shen YW, Boshnjaku V, et al. Folic acid remodels chromatin on Hes1 and Neurog2 promoters during caudal neural tube development. J Biol Chem 2010; 285:36922 - 36932
- Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol 2010; 28:1057 - 1068
- Yao TP, Oh SP, Fuchs M, Zhou ND, Ch'ng LE, Newsome D, et al. Gene dosage-dependent embryonic development and proliferation defects in mice lacking the transcriptional integrator p300. Cell 1998; 93:361 - 372
- Ruthenburg AJ, Li H, Patel DJ, Allis CD. Multivalent engagement of chromatin modifications by linked binding modules. Nat Rev Mol Cell Biol 2007; 8:983 - 994
- Bu P, Evrard YA, Lozano G, Dent SY. Loss of Gcn5 acetyltransferase activity leads to neural tube closure defects and exencephaly in mouse embryos. Mol Cell Biol 2007; 27:3405 - 3416
- Lin W, Zhang Z, Srajer G, Chen YC, Huang M, Phan HM, et al. Proper expression of the Gcn5 histone acetyltransferase is required for neural tube closure in mouse embryos. Dev Dyn 2008; 237:928 - 940
- Dunwoodie SL, Rodriguez TA, Beddington RSP. Msg1 and Mrg1, founding members of a gene family, show distinct patterns of gene expression during mouse embryogenesis. Mech Dev 1998; 72:27 - 40
- Bhattacharya S, Michels CL, Leung MK, Arany ZP, Kung AL, Livingston DM. Functional role of p35srj, a novel p300/CBP binding protein, during transactivation by HIF-1. Genes Dev 1999; 13:64 - 75
- Yin Z, Haynie J, Yang X, Han B, Kiatchoosakun S, Restivo J, et al. The essential role of Cited2, a negative regulator for HIF-1α, in heart development and neurulation. Proc Natl Acad Sci USA 2002; 99:10488 - 10493
- Barbera JP, Rodriguez TA, Greene ND, Weninger WJ, Simeone A, Copp AJ, et al. Folic acid prevents exencephaly in Cited2 deficient mice. Hum Mol Genet 2002; 11:283 - 293
- Finnell RH, Waes JGV, Eudy JD, Rosenquist TH. Molecular basis of environmentally induced birth defects. Annu Rev Pharmacol Toxicol 2002; 42:181 - 208
- Menegola E, Di Renzo F, Broccia ML, Prudenziati M, Minucci S, Massa V, et al. Inhibition of histone deacetylase activity on specific embryonic tissues as a new mechanism for teratogenicity. Birth Defects Res B Dev Reprod Toxicol 2005; 74:392 - 398
- Massa V, Cabrera RM, Menegola E, Giavini E, Finnell RH. Valproic acid-induced skeletal malformations: associated gene expression cascades. Pharmacogenet Genomics 2005; 15:787 - 800
- Okada A, Kushima K, Aoki Y, Bialer M, Fujiwara M. Identification of early-responsive genes correlated to valproic acid-induced neural tube defects in mice. Birth Defects Res A Clin Mol Teratol 2005; 73:229 - 238
- Eikel D, Lampen A, Nau H. Teratogenic effects mediated by inhibition of histone deacetylases: evidence from quantitative structure activity relationships of 20 valproic acid derivatives. Chem Res Toxicol 2006; 19:272 - 278
- Lagger G, O'Carroll D, Rembold M, Khier H, Tischler J, Weitzer G, et al. Essential function of histone deacetylase 1 in proliferation control and CDK inhibitor repression. EMBO J 2002; 21:2672 - 2681
- Montgomery RL, Hsieh J, Barbosa AC, Richardson JA, Olson EN. Histone deacetylases 1 and 2 control the progression of neural precursors to neurons during brain development. Proc Natl Acad Sci USA 2009; 106:7876 - 7881
- Vega RB, Matsuda K, Oh J, Barbosa AC, Yang X, Meadows E, et al. Histone deacetylase 4 controls chondrocyte hypertrophy during skeletogenesis. Cell 2004; 119:555 - 566
- Cheng HL, Mostoslavsky R, Saito S, Manis JP, Gu Y, Patel P, et al. Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc Natl Acad Sci USA 2003; 100:10794 - 10799
- Glozak MA, Sengupta N, Zhang X, Seto E. Acetylation and deacetylation of non-histone proteins. Gene 2005; 363:15 - 23
- Minucci S, Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer 2006; 6:38 - 51
- Kim JH, Saraf A, Florens L, Washburn M, Workman JL. Gcn5 regulates the dissociation of SWI/SNF from chromatin by acetylation of Swi2/Snf2. Genes Dev 2010; 24:2766 - 2771
- Portoso M, Cavalli G. Morris K. The role of RNAi and noncoding RNAs in Polycomb mediated control of gene expression and genomic programming. RNA and the regulation of gene expression: a hidden layer of complexity 2088:Caister Academic Press 29 - 44
- Schones DE, Cui K, Cuddapah S, Roh TY, Barski A, Wang Z, et al. Dynamic regulation of nucleosome positioning in the human genome. Cell 2008; 132:887 - 898
- Bultman S, Gebuhr T, Yee D, La Mantia C, Nicholson J, Gilliam A, et al. A Brg1 null mutation in the mouse reveals functional differences among mammalian SWI/SNF complexes. Mol Cell 2000; 1287 - 1295
- Banting GS, Barak O, Ames TM, Burnham AC, Kardel MD, Cooch NS, et al. CECR2, a protein involved in neurulation, forms a novel chromatin remodeling complex with SNF2L. Hum Mol Genet 2005; 14:513 - 524
- Kim JK, Huh SO, Choi H, Lee KS, Shin D, Lee C, et al. Srg3, a mouse homolog of yeast SWI3, is essential for early embryogenesis and involved in brain development. Mol Cell Biol 2001; 21:7787 - 7795
- Gyuris A, Donovan DJ, Seymour KA, Lovasco LA, Smilowitz NR, Halperin AL, et al. The chromatin targeting protein Brd2 is required for neural tube closure and embryogenesis. Biochim Biophys Acta 2009; 1789:413 - 421
- Euskirchen GM, Auerbach RK, Davidov E, Gianoulis TA, Zhong G, Rozowsky J, et al. Diverse roles and interactions of the SWI/SNF chromatin remodeling complex revealed using global approaches. PLoS Genet 2011; 7:1002008
- Wu M, Zhang Y, Wu NH, Shen YF. Histone marks and chromatin remodelers on the regulation of neurogenin1 gene in RA induced neuronal differentiation of P19 cells. J Cell Biochem 2009; 107:264 - 271