Abstract
DNA methylation has been associated with age-related disease. Intra-individual changes in gene-specific DNA methylation over time in a community-based cohort has not been well described. We estimated the change in DNA methylation due to aging for nine genes in an elderly, community-dwelling cohort of men. Seven hundred and eighty four men from the Veterans Administration Normative Aging Study who were living in metropolitan Boston from 1999–2009 donated a blood sample for DNA methylation analysis at clinical examinations repeated at approximately 3-5 year intervals. We used mixed effects regression models. Aging was significantly associated with decreased methylation of GCR, iNOS and TLR2 and with increased methylation of IFNγ, F3, CRAT and OGG. Obstructive pulmonary disease at baseline modified the effect of aging on methylation of IFNγ (interaction p = 0.04). For participants who had obstructive pulmonary disease at their baseline visit, the rate of change of methylation of IFNγ was -0.05% 5-methyl-cytosine (5-mC) per year (95% CI: -0.22, 0.13), but was 0.14% 5-mC per year (95% CI: 0.05, 0.24) for those without this condition. Models with random slopes indicated significant heterogeneity in the effect of aging on methylation of GCR, iNOS and OGG. These findings suggest that DNA methylation may reflect differential biological aging.
Keywords: :
Epigenetics has been defined as the study of any potentially stable and, possibly, heritable change in gene expression or cellular phenotype that occurs without changes in the base-pairing of DNA.Citation1 DNA methylation is the best studied of the epigenetic processes. Hypomethylation of regulatory sequences tends to correlate with increased gene expression, while increased methylation usually results in transcriptional suppression.Citation2
Epigenetic modifications in disease processes have been well studied. The largest body of literature is in the area of cancer research but such changes in the epigenome are increasingly being linked to other age-related diseases, such as Alzheimer diseaseCitation3,Citation4 and atherosclerosis.Citation5-Citation7 Epigenetic changes may also occur during normal aging. The relationship between aging and DNA methylation was originally proposed in a study of humpbacked salmon; since then, the association of decreased global DNA methylation and age has been supported in studies of mouse, rat, and human tissues.Citation8 In a study of twins varying in age from 3 to 74 y, global and locus-specific epigenetic differences between the pairs were found to increase with age;Citation9 however, the study did not follow the same individuals over time. Hypomethylation of repetitive elements (i.e., DNA sequences mostly located in intergenic regions) has also been associated with age in epidemiologic studies.Citation10,Citation11
We are aware of only one study that has reported changes in gene-specific methylation over time within the same individual.Citation12 This study examined both global and gene-specific methylation. While the population-average global DNA methylation did not change over time, there were intra-individual changes, with about the same number of individuals showing a decrease as an increase.Citation12 Gene-specific methylation in this study was analyzed in a subset of 41 individuals in the cohort and results were only presented for a family of five.
Given the lack of studies reporting on changes in DNA methylation in community-based cohorts in the literature, we set out to describe the intra-individual change in gene-specific methylation in a cohort of over 700 men. Within the context of an ongoing study, we used a candidate gene approach to examine DNA methylation. We selected the following genes for analysis: glucocorticoid receptor (GCR), inducible nitric oxide synthase (iNOS), intercellular adhesion molecule (ICAM), interleukin‑6 (IL-6), toll-like receptor‑2 (TLR2), interferon-gamma (IFN-gamma), coagulation factor-3 (F3), carnitine O‑acetyltransferase (CRAT), and 8‑oxoguanine DNA glycosylase 1 (OGG). These genes were chosen because they are expressed with variable degree in leukocytes,Citation13 the DNA source used in our study, and because of their relation to inflammation, endothelial function, and oxidative metabolism, processes known to be associated with aging and age-related diseases. We hypothesized that change in methylation over time would be dependent on the age of the participant at the baseline visit and wanted to distinguish between these two effects—cross-sectional age difference between subjects and change in age in each subject over time. We further hypothesized that the direction of change would vary by gene and by individual due to chronic conditions associated with age or other factors. We were able to test these hypotheses through secondary analyses of interaction and random effects models. Here, we describe the changes in methylation of these nine genes in a longitudinal cohort of elderly, community-dwelling men.
Results
The mean age of participants at baseline was 72 y (). Subjects had between one and five measurements of DNA methylation for each gene. describes the characteristics of each gene at each visit. The correlations between mean DNA methylation of each gene measure at the first and second visit were also calculated (). Correlations ranged from high for IL6 (rho = 0.705, p < 0.0001) to moderate for ICAM (rho = 0.557, p < 0.0001) to low for GCR (rho = 0.116, p = 0.0118).
Table 1. Baseline Characteristics of Participants in the Normative Aging Study, Boston, MA, 1999 - 2009 [mean (SD) or n (%)]
Table 2. Descriptive Statistics of Gene-specific Methylation by Visit for Participants in the Normative Aging Study, Boston, MA, 1999 - 2009
In the adjusted analysis between age at visit and DNA methylation for the 9 genes (Model 1), increased age was associated with increased DNA methylation for TLR2 at the p < 0.05 level (). Increased age at visit was also associated with decreased DNA methylation of iNOS at p < 0.05. Models with a term for the cross-sectional effect of age and a term for the longitudinal effect of age (Model 2) were examined next. After adjustment, there was a statistically significant association at the p < 0.05 level between DNA methylation and the longitudinal effect of age for many of the genes examined. For GCR, iNOS, and TLR2, age was associated with decreased methylation. For IFNγ, F3, CRAT, and OGG, age was associated with increased methylation (). The cross-sectional effect of age was statistically significantly associated with decreased DNA methylation of iNOS and increased DNA methylation of TLR2 in adjusted models. After restricting the sensitivity analysis to participants who did not have a diagnosis of IHD at baseline, the associations persisted, except for TLR2, which lost statistical significance but remained in the same direction for both the cross-sectional and longitudinal effect of age.
Table 3. Association Between Gene-specific Methylation and Age at Visit in the Normative Aging Study, Boston, MA, 1999 - 2009
Table 4. Association Between Gene-specific Methylation and Cross-sectional Age and Change in Age Over Time in the Normative Aging Study, Boston, MA, 1999 - 2009
A statistically significant interaction between obstructive pulmonary disease at baseline and change in age was found for methylation of IFNγ (interaction p = 0.04). For participants who had obstructive pulmonary disease at their baseline visit, the rate of change of methylation of IFNγ was -0.05% 5‑mC per year (95% confidence interval: -0.22, 0.13) but was 0.14% 5‑mC per year (95% confidence interval: 0.05, 0.24) for those without this condition. There was no statistically significant interaction between either diabetes mellitus or obesity at baseline and change in age for methylation for any of the genes investigated.
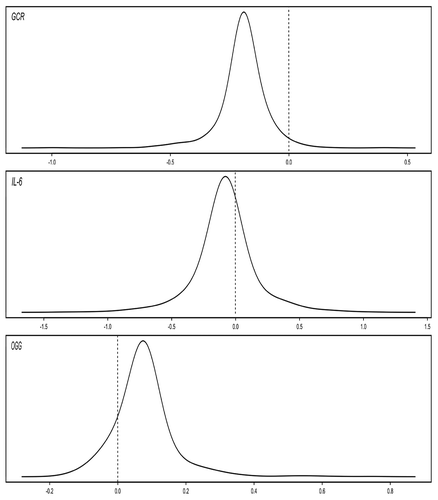
Finally, models with random intercepts and slopes for the effect of age were fit. For three of the genes evaluated (GCR, iNOS, and OGG), there was significant heterogeneity in the effect of change in age, indicating differing rates of change among subjects. displays the distribution of the subject-specific slope estimates for those genes.
Figure 1. Plots of Subject-specific Slopes for Change in Age in the Normative Aging Study, Boston, MA, 1999 – 2009.
Discussion
We examined changes in methylation patterns of nine genes over time in a cohort of elderly men. After adjusting for covariates, we found that change in age was significantly associated with decreased methylation of GCR, iNOS and TLR2 and with increased methylation of IFNγ, F3, CRAT and OGG. By estimating random slopes, we also found that this change was modified by unmeasured characteristics that varied between individuals in our population.
The association between global measures of DNA methylation and age has been previously studied in both animals and humans. Global loss of DNA methylation during aging has been reported in tissue from rats, mice, and cows.Citation14 Decreased genomic methylation has also been seen in human lymphocytesCitation15 and peripheral blood cells.Citation16 In a study examining methylation changes within the same individuals over time, Bjornsson et al.Citation12 found both losses and gains in global DNA methylation over time in different individuals, resulting in no change in the population average. Bollati et al. found that aging was associated with a decline in repetitive element methylation in the same cohort as we investigated here.Citation10
A number of human tissue studies have reported associations between cross-sectional age and gene-specific DNA methylation. Hypermethylation of a number of specific genes has been associated with cross-sectional age including the estrogen receptor (ER) (human colonic mucosa),Citation17 insulin-like growth factor II (IGF2) (colonic tissue),Citation18 p14 (colonic tissue),Citation19 p16 (gastric epithelia),Citation20 E-cadherin (urothelial bladder cells),Citation21 c‑fos (liver tissue),Citation22 and collagen α 1 (periodontal ligament tissue).Citation23 In addition, genes associated with colorectal cancer, including MYOD and N33, have been associated with age-related partial methylation in normal colon mucosa.Citation24 A study of over 200 human tissue samples from 10 anatomic sites also found a significant association between aging and DNA methylation.Citation25
The study by Bjornsson et al. is the only other study that we are aware of that has examined intra-individual gene-specific DNA methylation changes due to aging in a human cohort. Gene-specific methylation was analyzed in a subset of 41 individuals in their cohort, though they only reported the gene-specific methylation results for 50 genes that showed the greatest change over time in 5 members of a family. Though none of the genes we investigated were included in this group of 50, we saw both increases and decreases in methylation over time in our study, depending on the gene being analyzed.
When biological markers change over time in aging individuals, the markers will be associated with age at any given time. Therefore, a cross-sectional analysis will be influenced by selection of study participants because values for older participants will represent only those who survived up until that age.Citation26 A. Munoz and S.J. Gange, Methodological issues for biomarkers and intermediate outcomes in cohort studies, Epidemiol Rev. 1998; 20:29–42.Citation27 In this study, we were able to make use of repeated measurements of DNA methylation on a large sample of individuals. This allowed us to distinguish between the cross-sectional effect of age (i.e., baseline effect) and the longitudinal effect (i.e., effect of years since the first visit).
By examining the longitudinal effect, we found that aging was significantly associated with decreased methylation of iNOS, TLR2, and GCR. Methylation of iNOS has been shown to suppress iNOS expression.Citation28 While we cannot conclude anything about expression of iNOS in the present study, iNOS is associated with inflammatory processes and inflammation is common in aging and age-related disease. Similarly, age-associated defects in the innate immune system have been observed and human studies have found an association between aging and decreased TLR induced cytokine production. In a study of rhesus monkeys, glucocorticoid receptor genes were upregulated with aging.Citation29
We also found that aging was significantly associated with increased methylation of F3, CRAT, and OGG. Again, while we did not measure gene expression in our study participants, each of these genes has been associated with markers of aging and age-related diseases. Markers of activated coagulation, such as factor VIII,Citation30 as well as increased oxidative stress damage, have been shown to be increased with age. Carnitine acetyltransferase (CRAT) is a key enzyme in the metabolic pathway in mitochondria, peroxisomes and endoplasmic reticulum, and carnitine decline is a trait of an insulin resistant state associated with increasing age.Citation31 Overall, while our study cannot draw conclusions on how gene expression may relate to aging, the findings point to continued DNA methylation changes throughout the life course and indicate that DNA methylation may reflect differential biological aging. How these changes relate to gene expression requires further investigation.
We found a significant interaction between having obstructive pulmonary disease at baseline and change in age when looking at methylation of IFN‑γ . We also found significant heterogeneity between subjects for the longitudinal effect of age on methylation of three different genes—GCR, IL‑6, and OGG. Interestingly, we saw no effect of aging on the methylation of IL‑6 in our main analysis because methylation of this gene increased over time in some participants, and decreased over time in others. Taken together, these findings demonstrate that changes in methylation over time do not only vary by gene, but also by individual.
This study has several limitations. Methylation analysis was performed on white blood cell (WBC) DNA and it is not clear to what extent the changes we observed in WBC DNA reflect similar modification of DNA methylation in target tissue. However, all the genes we selected are known to be expressed and functional in WBCs, and participate in critical cellular functions in WBCs such as inflammation and oxidative metabolism. Therefore, measurements in WBCs may be appropriate biological markers.
There is still much uncertainty regarding factors that may influence DNA methylation, and, therefore, the causal pathway between aging and DNA methylation is not clearly defined. Our multivariate models included a number of covariates that are possible predictors of DNA methylation. While the effect estimates did not change substantially between the unadjusted and adjusted results, it is also possible that these covariates are intermediates in the mechanistic pathway between age and DNA methylation. If so, our adjusted analysis may be too conservative,
Our cohort consists of elderly, white males. While we found an association between aging and DNA methylation, it is important to note that our youngest subject was 55 y old at baseline. Therefore, it is not clear how aging would be associated with DNA methylation in a younger cohort or one with a wider age range. It is also not clear if changes in DNA methylation vary by gender.
Epigenetic mechanisms are increasingly being recognized as playing an important role in chronic disease. For the first time, the intra-individual change in DNA methylation of 9 genes has been described in a large, community-based cohort. The longitudinal nature of the study enabled examination of the effect of aging over time, while adjusting for cross-sectional age. This study also found that the effect of aging could vary by gene and individual, either due to known clinical conditions or other unidentified factors. These results have implications for understanding the links between epigenetics, aging, and age-related diseases.
Methods
Study population
This study included 784 elderly men who, as of March 1999, were active participants in the Normative Aging Study (NAS). The NAS cohort was established by the Veterans’ Administration in 1961, which enrolled men 21 to 80 y of age from the greater Boston, MA area who were free of known chronic medical conditions.Citation32 Since the time of enrollment, participants have had comprehensive clinical examinations at 3- to 5‑y intervals. Further details can be found elsewhere.Citation33 In examinations that took place between March 1999 and May 2009, 784 active participants agreed to donate at least one whole blood sample that was used for DNA methylation analysis. Because of assay failure and the limited amount of DNA available from each subject, DNA methylation analysis was successful on varying numbers of subjects for each gene ().
Exposure variable and covariates
NAS participants reported to the study center on the morning of their scheduled examinations. Age, height and weight were assessed at the visit. A physician elicited a complete medical history, including daily medications. Current smoking and past history of smoking were assessed using the American Thoracic Society questionnaire.Citation34
Methylation analysis
DNA was extracted from stored frozen buffy coat of 7 mL whole blood. DNA methylation analyses were performed on bisulfite-treated DNA using highly quantitative analysis based on PCR pyrosequencing; 500 ng DNA (concentration 25 ng/μL) was treated using the EZ DNA Methylation-Gold Kit (Zymo Research, Orange, CA, USA) according to the manufacturer’s protocol. Final elution was performed with 200 μL M-Elution Buffer. The assay for methylation was developed by locating the promoters using Genomatix Software (Genomatix Software Inc., Ann Arbor, MI) as described in Table S1. For each assay a 50 μL PCR was performed in 25 μL GoTaq Green Master mix (Promega), 25 ng bisulfite-treated genomic DNA, and water. PCR cycling conditions and reagent amounts are described in Table S2. PCR products were purified and sequenced by pyrosequencing as previously describedCitation35 using 0.3 μM sequencing primer. Primers and sequences analyzed for each assay are shown in Table S3. For each assay, primer sets and conditions were evaluated using a two-step approach. In the first step, correct PCR amplification was verified on standard DNA by running the PCR products on an agarose gel. The standard DNA was comprised of a pool of human DNA from healthy volunteers. In the second step, five different samples of normal DNA from healthy volunteers were run starting from bisulfite treatment through PCR amplification and Pyrosequencing to test the entire analytic pipeline. The mean CV was 0.02 for GCR, 0.02 for iNOS, 0.06 for ICAM, 0.02 for IL-6, 0.04 for TLR2, 0.002 for IFNγ, 0.08 for F3, 0.05 for CRAT, and 0.15 for OGG. Primer sets/conditions that did not produce successful pyrosequencing analyses, as indicated by the built-in Pyrosequencing software (Pyro Q‑CpG Software), were modified and retested. In this second step, all samples were run in duplicates. Primer sets and conditions with low reproducibility were modified and retested. DNA methylation analysis measured multiple individual CpG dinucleotide positions for each marker and was repeated twice on each sample to minimize the assay variability. The mean value of all dinucleotide positions was used in all statistical analyses.
Statistical analysis
Due to the repeated measures of DNA methylation for many participants, the association between age at visit and DNA methylation was evaluated with a mixed effects model (Model 1) for each gene, to account for the correlation among measurements within the same subject. The following was assumed:
1)
Yit = β0 + ui + β1 age + β2X2+ … + βpXp (Model
where Yit was the level of gene-specific methylation (measured as % 5‑methyl-cytosine) in subject i at time t, β0 was the overall intercept, and ui was the separate random intercept for subject i. In the above model X2–Xp are the covariates, which were included only in multivariable models.
In multivariable models, the following potential confounders or predictors of gene-specific methylation were chosen a priori based on previous literatureCitation10,Citation36and included in the analysis: body mass index (BMI), systolic blood pressure (SBP), diastolic blood pressure (DBP), fasting blood glucose (FBG), use of a statin medication, smoking status (never/former/current) and pack-years smoked, and season and day of week of blood draw. Percent Lymphocytes and % neutrophils were also included to control for potential age-related variation among leukocyte populations in gene-specific methylation.
The coefficient of age in model 1 reflects both the longitudinal change in each subject over time, as well as the cross-sectional difference between subjects, based on their age. If DNA methylation is associated with survival, the latter is likely to underestimate the true rate of change with time. To further explore these two different aspects, a second model for each gene (Model 2) was fit that included both: (i) age at the first visit (baseline age) and; (ii) years elapsed since the first visit (change in age). The following was assumed:
2)
Yit = β0 + ui + β1 baseline age + β2 change in age + β3X3+ … + βpXp (Model
where β1 was the coefficient to estimate the cross-sectional effect of age and β2 was the coefficient to estimate the longitudinal effect of age. The correlation between baseline age and change in age was rho = -0.17. The same covariates were included in Model 2 as were included in Model 1.
To determine if clinical characteristics that are prevalent in an aging cohort modified the association between aging and DNA methylation, models with interaction terms for such conditions at baseline were fit, as a secondary analysis. The following three clinical conditions were examined as potential effect modifiers: obesity (defined as BMI ≥ 30), obstructive pulmonary disease (defined as FEV1/FVC < 0.7), and diabetes mellitus (defined as physician-diagnosed diabetes mellitus or fasting blood glucose > 126 mg/dL). The regression models included cross-product terms for the interaction between those modifying factors and change in age, along with the main effects. If there was a significant interaction (at p < 0.05), effect estimates from each stratum were then calculated.
To determine if there was variation in the effect of aging between participants not captured by the specific clinical characteristics described, models with random intercepts and random slopes for the change in age were fit. These models allowed both the intercept and slope to vary randomly among subjects thereby allowing for heterogeneity of effect between subjects. A Wald test for the covariance of the random slopes was performed to determine if there was significant heterogeneity in the effect of age between subjects.
Finally, to determine if health status at baseline influenced the results, a sensitivity analysis was performed. Since DNA methylation has been associated with development of ischemic heart disease (IHD) in this cohort,Citation37 the main analysis examining the cross-sectional and longitudinal effects of age (Model 2) was repeated only on subjects who did not have diagnosed IHD at baseline (n = 551). IHD was defined as diagnosis with International Classification of Diseases, 9th Revision (ICD9) codes 410 – 414. All mixed effect models were conducted using the PROC MIXED procedure in SAS version 9.2 (SAS Institute, Cary, NC, USA). p values were two-sided.
| Abbreviations: | ||
| 5mC | = | 5-methylcytosine |
| CRAT | = | carnitine O-acetyltransferase |
| DNA | = | deoxyribonucleic acid |
| F3 | = | coagulation factor-3 gene |
| GCR | = | glucocorticoid receptor gene |
| iNOS | = | inducible nitric oxide synthase gene |
| ICAM | = | intercellular adhesion molecule gene |
| IFN-gamma | = | interferon-gamma |
| IL-6 | = | interleukin-6 gene |
| NAS | = | Normative Aging Study |
| TLR2 | = | toll-like receptor-2 gene |
| OGG | = | 8-oxoguanine DNA glycosylase 1 gene |
| PCR | = | polymerase chain reaction |
| VA | = | Veterans Administration |
| WBC | = | white blood cell |
Additional material
Download Zip (213.1 KB)Acknowledgments
This work was supported by the National Institue of Environmental Health Sciences at the National Institutes of Health (T32ES07069, ES0002, ES015172–01, ES014663, and T32ES016645). The VA NAS is supported by the Cooperative Studies Program/Epidemiology Research and Information Center of the US. Department of Veterans Affairs and is a component of the Massachusetts Veterans Epidemiology Research and Information Center, Boston, Massachusetts.
Related Research Data
REFERENCES
- Goldberg AD, Allis CD, Bernstein E. Epigenetics: A landscape takes shape. Cell 2007; 128:635 - 8; http://dx.doi.org/10.1016/j.cell.2007.02.006; PMID: 17320500
- Richardson B. Impact of aging on DNA methylation. Ageing Res Rev 2003; 2:245 - 61; http://dx.doi.org/10.1016/S1568-1637(03)00010-2; PMID: 12726774
- Gravina S, Vijg J. Epigenetic factors in aging and longevity. Pflugers Arch 2010; 459:247 - 58; http://dx.doi.org/10.1007/s00424-009-0730-7; PMID: 19768466
- Wang SC, Oelze B, Schumacher A. Age-specific epigenetic drift in late-onset alzheimer's disease. PLoS ONE 2008; 3:e2698; http://dx.doi.org/10.1371/journal.pone.0002698; PMID: 18628954
- Wierda RJ, Geutskens SB, Jukema JW, Quax PH, van den Elsen PJ. Epigenetics in atherosclerosis and inflammation. J Cell Mol Med 2010; 14:1225 - 40; http://dx.doi.org/10.1111/j.1582-4934.2010.01022.x; PMID: 20132414
- Turunen MP, Aavik E, Yla-Herttuala S. Epigenetics and atherosclerosis. Biochim Biophys Acta 2009; 1790:886-91.
- Pogribny IP, Beland FA. DNA hypomethylation in the origin and pathogenesis of human diseases. Cell Mol Life Sci 2009; 66:2249 - 61; http://dx.doi.org/10.1007/s00018-009-0015-5; PMID: 19326048
- Murgatroyd C, Wu Y, Bockmuhl Y, Spengler D. The janus face of DNA methylation in aging. Aging (Albany NY) 2010; 2:107 - 10; PMID: 20354272
- Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci USA 2005; 102:10604 - 9; http://dx.doi.org/10.1073/pnas.0500398102; PMID: 16009939
- Bollati V, Schwartz J, Wright R, Litonjua A, Tarantini L, Suh H, et al. Decline in genomic DNA methylation through aging in a cohort of elderly subjects. Mech Ageing Dev 2009; 130:234 - 9; http://dx.doi.org/10.1016/j.mad.2008.12.003; PMID: 19150625
- Jintaridth P, Mutirangura A. Distinctive patterns of age-dependent hypomethylation in interspersed repetitive sequences. Physiol Genomics 2010.
- Bjornsson HT, Sigurdsson MI, Fallin MD, Irizarry RA, Aspelund T, Cui H, et al. Intra-individual change over time in DNA methylation with familial clustering. JAMA 2008; 299:2877 - 83; http://dx.doi.org/10.1001/jama.299.24.2877; PMID: 18577732
- Aceview. http://www.ncbi.nlm.nih.gov/IEB/Research/Acembly/index.html
- Romanov GA, Vanyushin BF. Methylation of reiterated sequences in mammalian DNAs. effects of the tissue type, age, malignancy and hormonal induction. Biochim Biophys Acta 1981; 653:204 - 18; PMID: 7225396
- Drinkwater RD, Blake TJ, Morley AA, Turner DR. Human lymphocytes aged in vivo have reduced levels of methylation in transcriptionally active and inactive DNA. Mutat Res 1989; 219:29 - 37; PMID: 2911269
- Fuke C, Shimabukuro M, Petronis A, Sugimoto J, Oda T, Miura K, et al. Age related changes in 5-methylcytosine content in human peripheral leukocytes and placentas: An HPLC-based study. Ann Hum Genet 2004; 68:196 - 204; http://dx.doi.org/10.1046/j.1529-8817.2004.00081.x; PMID: 15180700
- Issa JP, Ottaviano YL, Celano P, Hamilton SR, Davidson NE, Baylin SB. Methylation of the oestrogen receptor CpG island links ageing and neoplasia in human colon. Nat Genet 1994; 7:536 - 40; http://dx.doi.org/10.1038/ng0894-536; PMID: 7951326
- Issa JP, Vertino PM, Boehm CD, Newsham IF, Baylin SB. Switch from monoallelic to biallelic human IGF2 promoter methylation during aging and carcinogenesis. Proc Natl Acad Sci USA 1996; 93:11757 - 62; http://dx.doi.org/10.1073/pnas.93.21.11757; PMID: 8876210
- Shen L, Kondo Y, Hamilton SR, Rashid A, Issa JP. P14 methylation in human colon cancer is associated with microsatellite instability and wild-type p53. Gastroenterology 2003; 124:626 - 33; http://dx.doi.org/10.1053/gast.2003.50102; PMID: 12612901
- So K, Tamura G, Honda T, Homma N, Waki T, Togawa N, et al. Multiple tumor suppressor genes are increasingly methylated with age in non-neoplastic gastric epithelia. Cancer Sci 2006; 97:1155 - 8; http://dx.doi.org/10.1111/j.1349-7006.2006.00302.x; PMID: 16952303
- Bornman DM, Mathew S, Alsruhe J, Herman JG, Gabrielson E. Methylation of the E-cadherin gene in bladder neoplasia and in normal urothelial epithelium from elderly individuals. Am J Pathol 2001; 159:831 - 5; http://dx.doi.org/10.1016/S0002-9440(10)61758-0; PMID: 11549575
- Choi EK, Uyeno S, Nishida N, Okumoto T, Fujimura S, Aoki Y, et al. Alterations of c-fos gene methylation in the processes of aging and tumorigenesis in human liver. Mutat Res 1996; 354:123 - 8; http://dx.doi.org/10.1016/0027-5107(96)00056-5; PMID: 8692198
- Takatsu M, Uyeno S, Komura J, Watanabe M, Ono T. Age-dependent alterations in mRNA level and promoter methylation of collagen alpha1(I) gene in human periodontal ligament. Mech Ageing Dev 1999; 110:37 - 48; http://dx.doi.org/10.1016/S0047-6374(99)00041-X; PMID: 10580690
- Ahuja N, Li Q, Mohan AL, Baylin SB, Issa JP. Aging and DNA methylation in colorectal mucosa and cancer. Cancer Res 1998; 58:5489 - 94; PMID: 9850084
- Christensen BC, Houseman EA, Marsit CJ, Zheng S, Wrensch MR, Wiemels JL, et al. Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CpG island context. PLoS Genet 2009; 5:e1000602; http://dx.doi.org/10.1371/journal.pgen.1000602; PMID: 19680444
- Ware JH, Dockery DW, Louis TA, Xu XP, Ferris BG Jr., Speizer FE. Longitudinal and cross-sectional estimates of pulmonary function decline in never-smoking adults. Am J Epidemiol 1990; 132:685 - 700; PMID: 2403109
- Muñoz A, Gange SJ. Methodological issues for biomarkers and intermediate outcomes in cohort studies. Epidemiol Rev 1998; 20:29 - 42; PMID: 9762507
- Chan GC, Fish JE, Mawji IA, Leung DD, Rachlis AC, Marsden PA. Epigenetic basis for the transcriptional hyporesponsiveness of the human inducible nitric oxide synthase gene in vascular endothelial cells. J Immunol 2005; 175:3846 - 61; PMID: 16148131
- Blalock EM, Grondin R, Chen KC, Thibault O, Thibault V, Pandya JD, et al. Aging-related gene expression in hippocampus proper compared with dentate gyrus is selectively associated with metabolic syndrome variables in rhesus monkeys. J Neurosci 2010; 30:6058 - 71; http://dx.doi.org/10.1523/JNEUROSCI.3956-09.2010; PMID: 20427664
- Conlan MG, Folsom AR, Finch A, Davis CE, Sorlie P, Marcucci G, et al. Associations of factor VIII and von willebrand factor with age, race, sex, and risk factors for atherosclerosis. the atherosclerosis risk in communities (ARIC) study. Thromb Haemost 1993; 70:380 - 5; PMID: 8259533
- Noland RC, Koves TR, Seiler SE, Lum H, Lust RM, Ilkayeva O, et al. Carnitine insufficiency caused by aging and overnutrition compromises mitochondrial performance and metabolic control. J Biol Chem 2009; 284:22840 - 52; http://dx.doi.org/10.1074/jbc.M109.032888; PMID: 19553674
- Bell B, Rose C, Damon A. The normative aging study: An interdisciplinary and longitudinal study of health and aging. Aging Hum Dev 1972; 3:4 - 17
- Park SK, O'Neill MS, Vokonas PS, Sparrow D, Schwartz J. Effects of air pollution on heart rate variability: The VA normative aging study. Environ Health Perspect 2005; 113:304 - 9; http://dx.doi.org/10.1289/ehp.7447; PMID: 15743719
- Ferris BG. Epidemiology standardization project (american thoracic society). Am Rev Respir Dis 1978; 118:1 - 120; PMID: 742764
- Bollati V, Baccarelli A, Hou L, Bonzini M, Fustinoni S, Cavallo D, et al. Changes in DNA methylation patterns in subjects exposed to low-dose benzene. Cancer Res 2007; 67:876 - 80; http://dx.doi.org/10.1158/0008-5472.CAN-06-2995; PMID: 17283117
- Zhu ZZ, Hou L, Bollati V, Tarantini L, Marinelli B, Cantone L, et al. Predictors of global methylation levels in blood DNA of healthy subjects: A combined analysis. Int J Epidemiol 2010.
- Baccarelli A, Wright R, Bollati V, Litonjua A, Zanobetti A, Tarantini L, et al. Ischemic heart disease and stroke in relation to blood DNA methylation. Epidemiology 2010; 21:819 - 28; http://dx.doi.org/10.1097/EDE.0b013e3181f20457; PMID: 20805753