Abstract
Drosophila melanogaster is a robust model to investigate many biological problems. It is however prone to some infections, which may endanger fly stocks if left unchecked for. One such infection is caused by an obligate fungal intracellular parasite, Tubulinosema ratisbonensis, which can be found in laboratory stocks. Here, we identify and briefly characterize a T. ratisbonensis strain that was infesting our Drosophila cultures and that required intensive measures to contain and eradicate the infection. We describe the phenotypes of infested stocks. We also report PCR-based techniques that allow the detection of infested stocks with a high sensitivity. We have developed a high-throughput qPCR assay that allows the efficient parallel screening of a large number of potentially-infested stocks. We also have investigated several prophylactic measures to prevent the further contamination of stocks, namely UV-exposure, ethanol treatment, bleaching, and desiccation. Bleaching was found to kill all spores. Other treatments were less effective but were found to be sufficient to prevent further contamination of noninfested stocks. Two treatments were efficacious in curing infested stocks (1) bleaching of eggs and subsequent raising of the larvae in clean vials; (2) fumagillin treatment. These cures only work on stocks that have not become too weak to withstand the procedures.
Introduction
Drosophila melanogaster is a major model in modern biology. One of the reasons for its success is the ease with which it can be cultured in the laboratory. Indeed, Drosophila stocks appear to be largely resistant to disease, thanks to an effective innate immune system.Citation1,Citation2 However, we have experienced in our laboratory an epidemic of unknown origin that afflicted many of our stocks grown at 25°C, but that apparently did not affect our stocks kept at 18°C. Expanded cultures of stocks suddenly collapsed and were rapidly lost, sometimes within a week. Several pathogens have been reported to infect fly cultures in the laboratory including viruses, bacteria, mycoplasma and spiroplasma, protozoa, nematodes and fungi.Citation3 Even though we did recover various bacteria and fungi from the hemolymph of infected flies, we identified microsporidia as being the primary cause of infection on the basis of consistent detection by light and electronic microscopy in diseased flies. Also, we did cause the disease by injecting spores into adult flies (Niehus et al., in preparation).
Microsporidia are highly derived fungi, which have an exclusively intracellular parasitic lifestyle.Citation4-Citation6 After going through several developmental stages, environmentally-resistant microsporidial spores generally burst out of infected cells and are released in the milieu following the demise of the host. An alternative to this horizontal mode of transmission is vertical transmission through the eggs in invertebrates.Citation7-Citation9 The mode of microsporidial penetration inside cells is highly original and is not fully understood. It involves the explosive extrusion of their polar tube into or next to a target cell. Next, the sporoplasm is ejected through the polar tube and may either find itself directly into the host cell cytoplasm or is subsequently phagocytosed. Alternatively, the spores may themselves be ingested by phagocytic cells, with the parasite subsequently escaping from the phagolysosome through its polar tube.Citation5 It appears that Drosophila larvae get infected by feeding on infected cadavers or possibly empty eggshells.Citation10,Citation11 The exact route of infection in the digestive tract remains to be established.
Of note, microsporidial infections are not limited to invertebrates but also occur in fishes and higher vertebrates, including immunodeficient patients.Citation12-Citation14 Microsporidia have rarely been reported to infect Drosophila.Citation10,Citation15-Citation19 Yet, it is likely that many laboratories working on Drosophila have experienced microsporidia infections and have not reported these in scientific publications. Indeed, reports can be found on the Internet, e.g., http://labs.csb.utoronto.ca/godt/homepage%20links/Microsporidia%20page.html and http://www.bio.net/bionet/mm/dros/2004-July/006629.html. Here, we report cell and molecular biology techniques that allow the identification and high-throughput detection of the strain of microsporidia that has infested our cultures. Finally, we report on efficacious prophylaxis measures and on a strategy to cure the infection.
Results and Discussion
Identification of the infecting species of microsporidia
The infested adult flies appeared to be large and displayed a swollen or sometimes distended abdomen (). We identified yeast-like microorganisms in our flies using both light and electron microscopy (). One of them had actually extruded its polar tube, which led us to the tentative identification of the infective agent as a microsporidium. Indeed, we did detect Uvitex 2B (chitin stain) positive signals using the technique described in Franzen et al. (; see also Materials and Methods section).Citation15 We therefore amplified by PCR a portion of the 18S rRNA of the parasite using primers (ss530f and ls580r) designed to hybridize with conserved regions of microsporidial rRNA (). Subsequent sequencing of the cloned 1475 nucleotide-long PCR fragment allowed us to identify this strain as being highly related to Tubulinosema ratisbonensis, with only two nucleotides being different out of 1475 for which the alignment could be made (). A multiple alignment of 1475 nucleotides between our strain, the published T. ratisbonensis sequence and that of T. kingi is shown in . Indeed, as previously described, T. ratisbonensis appears to be phylogenetically closely related to another Drosophila parasite, T. kingi and somewhat less to the mosquito parasite Anncaliia algerae (). In contrast, it does not belong to the branch that comprises vertebrate microsporidia species such as Encephalitozoon cuniculi, Enterocytozoon bieneusi and the bee parasites Nosema apis and N. ceranae (T. kingi used to be classified in the Nosema genus).
Figure 1. Macroscopic and microscopic detection of microsporidial infestation in adult Drosophila flies. (A) Comparison of noninfested female (left upper panel) to microsporidia-infested adult female (right upper panel), and noninfested male (left lower panel) to infested adult male (right lower panel). (B) Uvitex 2B staining (chitin stain) on fly smears of noninfested (upper panel) and highly infested (lower panel) adult flies, respectively. Arrows point to some mature spores examined microscopically either with bright field (left panels) or under fluorescence (right panels). (C) A typical microsporidial spore stage as observed by TEM in the cytoplasm of the cells of fat body lobules of a highly infested adult fly. PF, Polar filament; EX, exospore; N, nucleus (note that the Nosema and Tubulinosema genera are characterized by diplokaryotic nuclei). Scale bar: 500 nm.
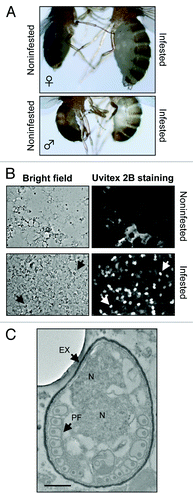
Figure 2. Sequence of the rDNA locus of the parasite infesting Drosophila stocks in the Strasbourg laboratory. Primer pairs used for different PCR techniques are shown within the identified 1475 bp large rRNA fragment of T. ratisbonensis (Strasbourg).

Figure 3. Alignment of the rDNA sequence of the Strasbourg parasite to Drosophila-infesting Tubulinosema species. This figure shows the rDNA sequences of the Strasbourg strain (T. rat. [Stras.]), of T. ratisbonensis (T. rat. [Regen.]), and of T. kingi. The lower sequence represents the consensus. Sequence divergence is indicated by bold nucleotides. N, unresolved nucleotide.
![Figure 3. Alignment of the rDNA sequence of the Strasbourg parasite to Drosophila-infesting Tubulinosema species. This figure shows the rDNA sequences of the Strasbourg strain (T. rat. [Stras.]), of T. ratisbonensis (T. rat. [Regen.]), and of T. kingi. The lower sequence represents the consensus. Sequence divergence is indicated by bold nucleotides. N, unresolved nucleotide.](/cms/asset/c41b619b-5488-41e7-a032-f491cb1c2cec/kfly_a_10920896_f0003.gif)
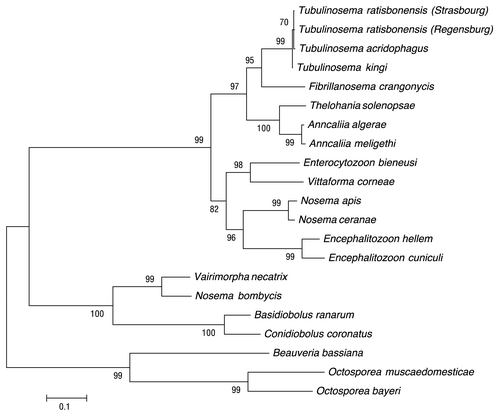
Figure 4. Phylogenetic analysis of various microsporidial species. Maximum composite likelihood analysis of small subunit rDNA showing the relationships of the identified microsporidia species in our fly cultures to other microsporidia. This analysis reveals the close relationship between T. ratisbonensis, T. kingi, and T. acridophagus. Bootstrap values are shown.
Detection of microsporidia
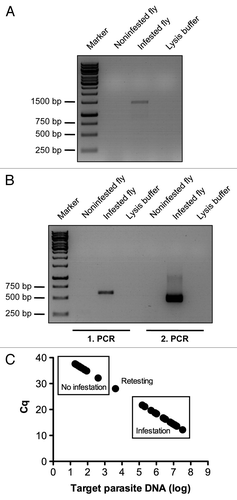
There are several methods to determine whether Drosophila are infested by microsporidia. Flies that are heavily infested displayed swollen abdomens as shown in , which most likely are distended because they contain a very high titer of microsporidia. However, these are rather extreme cases and a rampant infestation often goes by undetected until the stock suddenly collapses. A quick assay to assess whether a fly is infested is to make a smear of a fly on a microscopic slide and to stain it with a chitin-binding dye such as calcofluor or Uvitex 2B, which stains the microsporidial spore cell wall (). However, this method is impractical when having to test many potentially infected stocks. We therefore developed PCR-based assays, using either classical PCR () or nested PCR (), which is extremely sensitive but yields often false positives if the conditions are not perfectly controlled. We found that quantitative PCR (qPCR) () was a sensitive technique that was easily scaled up for high throughput detection of multiple Drosophila strains using 96-well plates for extraction of the samples and 96- or 384-well plates for performing the qPCR reactions.
Figure 5. Detection of microsporidial infestations of fly cultures by PCR techniques. Conventional PCR with universal microsporidia primer pair ss530f and ls580r (A) and nested-PCR (B) on crude gDNA extract of respectively noninfested and infested single adult flies. Lysis buffer used for gDNA extraction was used as a negative control for the PCR reaction (A and B). Large-scale detection of microsporidia infestation of fly cultures by qPCR is illustrated by the analysis of 40 randomly selected fly lines (C). Filled circles correspond to the PCR signal detected in single flies. The frames display the status of microsporidial infestation of tested fly lines as determined by qPCR. Note the presence of a line of undefined status in which the Cq is close to but less than 30. This line requires retesting. In practice, several individual flies are tested per fly line. All primer pairs used for analysis are listed in and their position within the cloned microspodial rRNA gene fragment from our infested fly cultures is illustrated in .
Prophylaxis of microsporidial infections
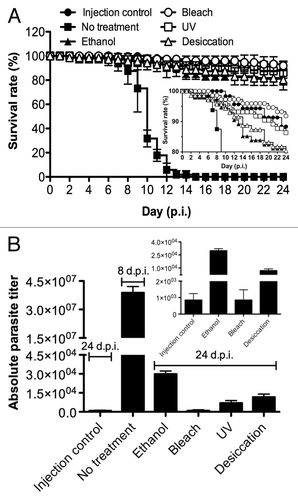
An essential step to ensure prophylaxis is to identify the treatments that are able to eradicate spores that might contaminate surfaces and tools that have been in contact with infected flies. To this end, we tested several disinfectants including 70% ethanol, UV, bleach treatments, and also desiccation on a surface to mimic an infected droplet drying on a bench. The outcome of these prophylactic measures was assessed both using fluorescence microscopy () and qPCR () following the infection of human cultured cells with the treated spore solution. Alternatively, the treated spore solutions were injected into flies and survival was then monitored (). Some spores could still be detected by Uvitex 2B staining two weeks after infection of the human fibroblasts, a majority of which may actually correspond to killed spores (). Because it is extremely difficult to extract gDNA from T. ratisbonensis spores with the procedure we have used (data not shown), the DNA that is amplified by qPCR actually corresponds to DNA extracted from intracellular proliferative stages, namely meronts and sporonts. While human cultured cells infected with nontreated spores yielded a large quantity of microsporidial amplifiable DNA, no signal was observed after bleach-treatment of the spores. The least effective treatment, UV, produced about 10% of the signal measured in the control. Desiccation and ethanol treatment still allowed some proliferation of the parasite, corresponding to 1–2% of the control (). Interestingly, all techniques were efficient enough to prevent killing flies injected with the treated spore solutions (). In keeping with the cell culture results, we did not detect any proliferation of the injected spores after bleach treatment 24 d after the injection (0.002% of control), but did observe some weak proliferation after ethanol (0.08% of control), UV (0.02% of control), and desiccation (0.03% of control) ().
Figure 6. Test of the efficiency of disinfection treatments using a cell culture assay. T. ratisbonensis spores were pretreated with 70% ethanol, 2,5% bleach, UV, or desiccated by drying on a surface (A). Cell monolayers of human lung fibroblasts (MRC-5) were infected with spores pretreated by these prophylaxis measures and cultured for two weeks at 31°C, with noninfected cells and nontreated infected cells as controls. Parasitic growth as revealed by the formation of foci (observed here only in the no treatment control) was examined by fluorescence microscopy after Uvitex 2B staining. Scale bar: 50 µm. (B) The absolute parasite titer was determined by qPCR. The blow-up graph illustrates the low parasite titer values obtained after ethanol and bleach treatment at low titers. Values shown represent the means +/− the standard deviations of biological triplicate samples. Each panel is representative of three independent experiments.
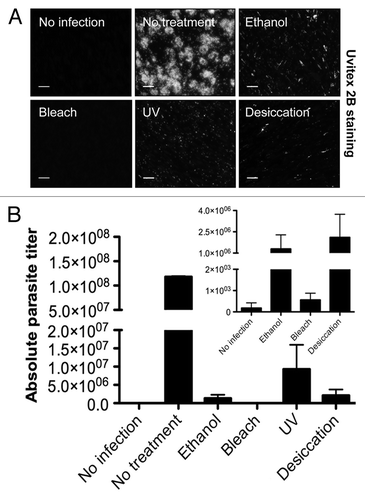
Figure 7. Test of the efficiency of disinfection treatments using an in vivo infection model. T. ratisbonensis spores were pretreated with 70% ethanol, 2,5% bleach, UV, or desiccated by air drying on a surface. Spore treated by these prophylaxis measures were injected into the body cavity of adult flies, with the injection of nontreated spores or buffer as respectively positive and negative controls. (A) Survival analysis: survival was monitored daily. The survival experiment shown is representative of at least six independent survival experiments. Values shown represent the means +/− the standard deviations of triplicate batches of each 20–25 flies hold at 29°C. The inset shows a blow-up of the curves between 80–100% survival. Error bars have been omitted for the sake of clarity. (B) The absolute parasite titer of flies shown in panel A was determined 24 d p.i., except for flies infected with untreated spores that were harvested 8 d p.i.. The blow-up graph shows the low parasite titer values obtained after bleach treatment. Values represent the means +/− the standard deviations of biological triplicate samples of five flies per treatment condition. The experiment shown is a representative of three independent experiments.
Treatment of infested stocks
When the stock is not too heavily infested and that females still lay eggs, we found that the most efficient way to rid the Drosophila stocks of the microsporidia is to dechorionate the eggs through bleaching as suggested by Futerman et al. for T. kingi.Citation10 In this manner, microsporidia spores deposited on the chorion of the eggs, which thus propagate the infection, are washed away and killed. It is important to check that the stock is effectively cleared. If not, a second round of dechorionation may be required at the next generation.
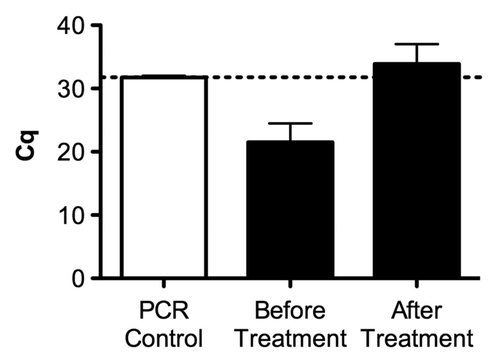
We have also tested the antimicrosporidial drug fumagillin, which targets methionine aminopeptidase2.Citation20 The drug was added to the fly food into culture tubes. Infested cultures of flies were allowed to oviposit on fumagillin containing fly food for some 12 h and removed afterwards. As shown in , the treatment was effective in curing infected flies as the measured titer using qPCR in treated offspring flies was undistinguishable from background signal.
Figure 8. Assessment of the efficiency of fumagilin treatment against microsporidia infestation as assessed by quantification of the infestation in fly stocks. The graph illustrates the microsporidia infestation of a representative fly stock before and after fumagillin treatment according to the procedures described in the material and method section. The success of the treatment was validated by qPCR: a low Cq value corresponds to a high dose of the parasite whereas a high Cq (> 30) may correspond to noninfested flies. The dotted line refers to the PCR control without DNA template. Values represent the means +/− the standard deviations of a biological duplicate.
Conclusions (Identification/Dissemination/Prophylaxis/Treatment)
Here, we have reported on the identification, detection, and prophylaxis/cure of microsporidial infections with T. ratisbonensis. Of note, in contrast to the previous report by Franzen et al., we found that infested flies displayed an external phenotype shown in . The reason for the discrepancy is not obvious and may be related either to the temperature of which the flies were reared or to the level of infection in each laboratory, i.e., we cannot exclude that our Drosophila cultures were more severely infested than those in Regensburg where T. ratisbonensis was first isolated. Of note, we have observed this phenotype in artificially infected flies, even when injected at the lowest dose (103 spores/fly), although only toward the end of the infection when flies were about to succumb (Niehus et al., in preparation).
Having identified the parasite that infested our Drosophila cultures, we have described several techniques to detect microsporidial infection by T. ratisbonensis varieties, which is likely rampant in many laboratories. One should suspect a microsporidial infection when flies display the phenotype shown in concomitant with the stock suddenly crashing within a week when expanded at 25°C. Actually, we have found that experimental infections are highly dependent on the temperature, with 18°C being a nonpermissive temperature (Niehus et al., in preparation). Of note, the nested-PCR and qPCR primers should also allow to amplify T. kingi DNA. The qPCR is the method of choice for the diagnosis of large numbers of fly cultures to identify microsporidia infestations. The advantages of qPCR over conventional PCR and nested-PCR are primarily the speed in which samples are processed (e.g., no need for agarose gel electrophoresis with intercalating fluorescent DNA dyes), the lower amounts of starting material needed for analysis, and its sensitivity to quantify the target DNA of interest. In our hands, the limit of sensitivity using our couple of primers (QnosemaF and QnosemaR) was about 10 molecules of the plasmid we use to standardize our assay [quantification cycle (Cq) of about 32]. We have defined a gray zone of uncertainty when measuring other pathogens, which corresponds to a Cq around 30. In case of doubt, a way to increase sensitivity would be to let flies age at 25°C before performing the analysis
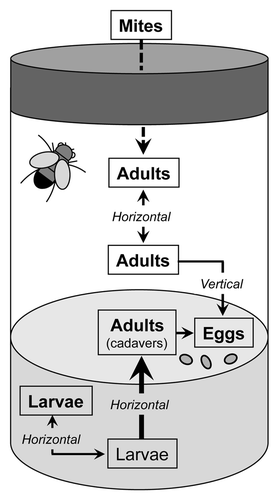
We find that bleach treatment is the most effective way to eradicate spores, even though some fish microsporidia have been reported to be highly resistant to chlorine treatment used to disinfect fish eggs.Citation21 Of note, the other techniques may be sufficient to prevent the spreading of the infection as a contamination in normal conditions would have to be at the larval stage through the oral route, which is much less efficient than through the direct injection into the hemocoel that we used here. Indeed, successful infestation of larvae has been achieved so far using high titers of spores (2.5 × 106 spores per 50 larvae at 20°C).Citation10 Thus, it appears that the spreading of the infection from vial to vial is most likely achieved by a vector, namely mites, and not by contamination of fly pads as the spores would desiccate (). As an extra precautionary step, we nevertheless cleaned our fly pads with bleach and treat with ethanol our brushes. Clearly, the first effective measure to prevent the spreading of the microsporidial epidemics is to eradicate mite infestation in cultured stocks.Citation3
Figure 9. Hypothetical route of infection in fly culture vials. Mites may likely serve as passive carriers for microsporidial spores, thus vectoring the infestation. Indeed, since spores are not resistant to desiccation, contamination on fly sorting pads or using contaminated equipment appears less likely. The main route of infection within a culture tube is likely the oral ingestion of spores by larvae feeding on infected adult cadavers. The horizontal transfer of spores excreted by feces from adult to adult, and a vertical transmission from mother to the offspring has been described experimentally with T. kingi, a close relative of T. ratisbonensis. As bleach treatment appeared to be an effective means to cure stocks of T. ratisbonensis, vertical transmission might not occur with this species. A horizontal transmission between larvae is likely occurring.
To cure stocks of the infection, we have attempted to make large-scale dechorionation of our stocks. This should be an effective procedure as eggs are exposed to bleach. We did not observe any sign of vertical transmission after dechorionation, which removes any spore deposited on the egg. However, this is labor-intensive and may result in the mixing up of stocks. An alternative is to use fumagillin treatment, but this could not be used on flies too weak to withstand the procedure because they were too heavily infested. In addition, the treatment may have to be applied twice for some heavily infested stocks. Another issue is that of toxicity. While fumagillin treatment is used in some parts of the world to cure honey bees of microsporidiosis, some toxicity has been observed in vertebrates. Also, fumagillin is a metabolite of the opportunistic pathogen A. fumigatus, and thus it may act as virulence factor. Indeed, it has been reported to impair the cellular immune response of lepidopteran larvae.Citation22 Finally, even though the treatment may appear safe in wild-type flies, it may not be the case for mutant stocks. Because we have not addressed thoroughly the issue of toxicity of this compound to the flies, it may be safer to use this treatment as a last resort
Once the Drosophila cultures have been cleaned from microsporidial infestation, the next step is to ensure that microsporidia no longer enter the laboratory. Thus, we have added in our quarantine procedure a step in which incoming stocks are tested for the presence of Wolbachia and of microsporidia using the qPCR assay we have described here.
MATERIAL AND Methods
Fly strains
Fly cultures were raised on standard cornmeal agar medium consisting of 6.4% (w/v) cornmeal (Moulin des moines, France), 4.8% (w/v) granulated sugar (Erstein, France), 1.2% (w/v) yeast, brewer’s dry powder (VWR, Belgium), 0.5% (w/v) agar (Sobigel, France) including 0.004% (w/v) 4-hydroxybenzoate sodium salt (Merck, Germany) at 25°C for propagation. The Exelixis w118 strain A5001 was used as a wild-type stock. Fly cultures used for experiments were routinely tested for microsporidia infection as described below by qPCR.
Cell and parasite culture
The human fibroblast MRC-5 cell line (kindly provided by Dr. Franzen, University of Regensburg, Germany) was used as a host cell line and maintained in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% (v/v) fetal calf serum (FCS), 1% (v/v) GlutaMAX (Invitrogen), 1% (v/v) PenStrep (Invitrogen), and 1 mM sodium pyruvate (Invitrogen). T. ratisbonensis (initially kindly provided by Dr. Franzen, University of Regensburg, Germany) was propagated in the MRC-5 cell line and cultured in DMEM supplemented with 2% (v/v) fetal calf serum (FCS), 1% (v/v) GlutaMAX (Invitrogen), 1% (v/v) PenStrep (Invitrogen), and 1 mM sodium pyruvate (Invitrogen). T. ratisbonensis spores were isolated and purified from host cells as described elsewhere.Citation23
Identification of microsporidia in fly cultures: DNA isolation, PCR amplification, cloning and DNA sequencing
Microsporidial genomic DNA (gDNA) was isolated from single adult flies showing macroscopic signs of microsporidia infestation (swollen and distended abdomen; ). Briefly, single flies were crushed with the help of a pestle in 50 µl Tris-buffer (10 mM Tris-HCl, pH 8; 1 mM EDTA; 25 mM NaCl) containing 2 mg/ml proteinase K (Qiagen) in a 1.5 ml reaction tube and then incubated for 1 h at 56°C, followed by heat-inactivation of proteinase K for 15 min at 70°C to obtain a crude extract of both fly and parasite gDNA. Routinely, 2 µl of crude gDNA extract was used for PCR amplification as template utilizing the GoTaq® Flexi DNA polymerase (Promega) according to the standard recommendations of the manufacturer. The conditions for PCR (final volume 50 µl) were as follows: initial activation was performed at 94°C for 2 min followed by 36 cycles of a three-step PCR. DNA strand separation was achieved at 94°C for 30 sec followed by annealing at 53°C for 60 sec, and extension at 72°C for 90 sec; a final extension was performed at 72°C for 5 min. The PCR amplification revealed an amplicon of 1475 bp, which includes part of the small subunit (SSU)-rRNA, the entire internal transcribed spacer (ITS) region, and part of the (large subunit) LSU-rRNA; it was amplified using the universal microsporida primer pair ss530f and ls580r [ss: primer in the small-subunit rRNA gene; ls: primer in the large subunit rRNA gene; f: forward primer (positive strand); r: reverse primer (negative strand)].Citation24 The sequence of the primer pair and their position within the cloned microspodial rRNA gene fragment from our infested fly cultures is illustrated in . The PCR product was loaded onto a 1% agarose gel containing ethidium bromide ().
Alignments
Alignments between parts of the rRNA genes of T. ratisbonensis (AY695845), T. kingi (DQ019419) and the amplicon (herein named as Strasbourg) obtained from our infested fly cultures by using the universal microsporidia primer pair ss530f and ls580r were created using MULTALIN.Citation25
Ultrastructural investigation of infested fly cultures: Transmission electron microscopy
For ultrastructural investigations, abdomens of microsporidia-infested flies were opened in PBS containing 4% glutaraldehyde and subsequently incubated for 30 min at RT in the same fixative. Afterwards, midguts were post-fixed at 4°C for 4 h with PBS containing 1% osmium tetroxide at RT, rinsed, dehydrated through a graded ethanol series, and embedded in epon/araldite resin. Ultra-thin sections were contrasted with uranyl acetate and lead citrate. Sections were investigated at 60 kV on a Hitachi 7500 transmission electron microscope.
Phylogenetic analysis
The evolutionary history was inferred using the Minimum Evolution method (ME).Citation26 The bootstrap consensus tree inferred from 500 replicates is taken to represent the evolutionary history of the taxa analyzed.Citation27 Branches corresponding to partitions reproduced in less than 50% bootstrap replicates are collapsed. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (500 replicates) are shown next to the branches.Citation27 The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The evolutionary distances were computed using the Maximum Composite Likelihood method and are in the units of the number of base substitutions per site.Citation28 The ME tree was searched using the Close-Neighbor-Interchange (CNI) algorithm at a search level of 0.Citation29 The Neighbor-joining algorithm was used to generate the initial tree.Citation30 The analysis involved 21 nucleotide sequences (). There were a total of 2732 positions in the final data set. Evolutionary analyses were conducted in MEGA5.Citation31
Table 2. Microsporidia species used to draw up the rDNA-based phylogenetic tree in
Microscopic detection of microsporida infestation: light- and fluorescence microscopy
The microscopic observation of microsporida-infected flies was conducted on fly smears from single adult flies. Briefly, single flies were smashed between two glass slides and spread uniformly on a glass slide. These fly smears were stained with 1% Uvitex 2B in PBS for 5 min at RT, washed carefully with ultrapure water (RiOS™, Millipore), then air-dried, and subsequently observed using a Axiovert 200M (Zeiss) inverted microscope equipped with a ApoTome device and a 63 × Plan Apochromat oil immersion objective.
Detection of microsporidia infestation: nested-PCR
gDNA was extracted from single flies by maceration as described above. Routinely, 2 µl of crude gDNA extract was used to perform the 1st PCR reaction (final volume 50 µl) as follows: initial activation was performed at 95°C for 2 min followed by 36 cycles of a three-step PCR. DNA strand separation was achieved at 95°C for 30 sec followed by annealing at 58°C for 40 sec, and extension at 72°C for 40 sec; a final extension was performed at 72°C for 3 min. The 1st PCR amplification revealed an amplicon of 561 bp. Subsequently, 2 µl of this amplicon served as a template for the 2nd PCR reaction (final volume of 50 µl) that was performed using the same PCR conditions, which revealed an amplicon of 450 bp. Primer pairs used are listed in .
Table 1. Sequences of primer pairs used for different PCR techniques
Detection of microsporidia infestation in fly cultures: Quantitative PCR
Large-scale routine tests for microsporidia infestations of fly cultures were performed on single adult flies by crushing flies (usually as multiple biological replicates) by a multiple 96-well homogenizer (Burkard Scientific) in 50 µl Tris-buffer (10 mM Tris-HCl, pH 8; 1 mM EDTA; 25 mM NaCl).Citation32 Alternatively, gDNA from single adult flies was extracted as described above. Routinely, 1 µl of crude gDNA extract was used for qPCR. PCR reactions (final volume 10 µl) were set up using iQ™ SYBR Green Supermix (BIO-RAD) according to the instructions of the manufacturer. Real-time PCR was then performed in 96-well plate format with an iCycler iQ™ Real Time detection system (BIO-RAD) or in 384-well plate formrat by using a CFX384 Real-Time PCR detection system (BIO-RAD). The conditions for qPCR were as follows: initial activation was performed at 98°C for 15 sec followed by 35 cycles. DNA strand separation was achieved at 95°C for 2 sec followed by an annealing at 60°C for 30 sec. Afterwards, a melting curve was generated ranging from 65°C to 95°C with an increment of 0.5°C each 5 sec. These melting curves were then analyzed for each data point to verify the specificity of the PCR reactions. Primers used are given in (QnosemaF and QnosemaR). Absolute quantification of parasite DNA levels was achieved using a standard ladder made with dilutions of a plasmid of known concentration and length, which contains the target sequence of interest (see below). The efficiency of the primers was measured on these standard curves and was usually about 100%. Only experiments in which the efficiency was over 85% were validated.
Plasmid standard for qPCR
The sequence of interest (part of rRNA gene of T. ratisbonensis) was amplified with the following primer pair: forward 5′-TCCCTGTTCTTTGCACACAC-3′ and reverse 5′-TGCATTACCAAGCAAGGCTA-3′. The conditions for PCR were as follows: initial activation was performed at 95°C for 2 min followed by 35 cycles of a three-step PCR. DNA strand separation was achieved at 95°C for 30 sec followed by annealing at 60°C for 30 sec, and extension at 72°C for 60 sec; a final extension was performed at 72°C for 5 min. The PCR product was purified after gel electrophoresis by PCR cleanup (NucleoSpin® Extract II; Nagel) and then ligated into the pGEM®-T Easy vector (Promega) according to manufacturer instructions. Plasmid transformation was conducted by using heat-shock transformation into Escherichia coli DH5 α. The plasmid was checked by sequencing.
Treatment of spores with different disinfectants conditions
A pellet of 5 × 107 T. ratisbonensis spores was treated either with 15 µl of 70% ethanol for 10 sec followed by the addition of 150 µl ultrapure water (RiOS™, Millipore), or 100 µl 2.5% bleach (sodium hypochlorite) in ultrapure water (RiOS™, Millipore) for 20 min, or five times UV (UV) irradiation at 300.000 µJ/cm2 (UV Stratalinker® 1800, Stratagene), respectively. Alternatively, spores were dried up on a surface and subsequently resuspended in a 100 µl drop of PBS. After treatment spores were centrifuged (3000 rpm, 10 min, RT), washed twice with sterile PBS, and resuspended in sterile PBS containing 0.01% Tween 20. They were then separated into aliquots of 10 µl containing a dose corresponding to 104 spores per 9.2 nl of the nontreated original solution. Untreated spores were mock treated (incubation with sterile PBS, including subsequent washing steps) and used as controls (aliquots of 10 µl containing 104 spores per 9.2 nl). PBS containing 0.01% Tween 20 was used as injection control. All treatments were conducted at room temperature (RT) under a laminar flow hood.
Assessment of the effectiveness of the disinfectant treatments in cell culture
MRC-5 cells were grown in 24-well plates to confluence and infected with spores that had undergone different disinfectant treatment conditions (see above). Culture medium was renewed after 7 d post infection (p.i.) to 50%. Cells used for qPCR were harvested by scrapping at 14 d p.i., followed by extraction of gDNA with the NucleoSpin® Tissue kit (Macherey-Nagel) according to manufacturer instructions. qPCR was performed as described above. Cells used for microscopy were fixed with 4% paraformaldehyde in PBS for 15 min at RT, washed once with PBS, then stained with 1% Uvitex 2B, washed three times with PBS, and directly observed in the 24-well using an Axiovert 200M (Zeiss) inverted microscope with a 10 × objective.
Assessment of the effectiveness of the disinfectant treatments in adult flies
Survival experiments were conducted as biological triplicates in batches of 20–25 wild-type flies (A5001) per vial. Treated spores (either ethanol, bleach, UV, or desiccation) were injected (9.2 nl of spore solution per fly, about 104 spores) into the thorax of adult flies with a micro capillary using the Nanoject II micro injector apparatus (Drummond, USA). Non-treated and treated spores were resuspended in PBS containing 0.01% Tween 20. PBS containing 0.01% Tween 20 was used as a control for injections. The vials containing the challenged flies were kept in an incubator at 29°C and surviving flies were counted each day. Flies were collected to determine the absolute parasite titer at 8 d.p.i. (no treatment injection control) and 24 d.p.i. for all other treatment conditions including the injection control. To this end, samples of at least five adult flies were frozen in liquid nitrogen and crushed with a Mixer Mill 300 (Retsch) twice for 60 sec at 20 Hz with 3 mm Tungsten Carbide Beads (Qiagen). Total gDNA was then prepared using the NucleoSpin® Tissue kit (Macherey-Nagel) according to the instructions of the manufacturer. qPCR was performed as described above.
Treatment of microsporidia infested fly cultures with Fumidil B
Infested fly cultures were cured from microsporidia infection with Fumidil B [the brand name for fumagillin (Thorne, UK)]. Fumidil B was mixed to viscous lukewarm fly food at a final concentration of 1 mg/ml. The F1 generation was tested for the success of the treatment by qPCR some 14 d after adult flies have hatched.
| Abbreviations: | ||
| SSU | = | small subunit rRNA |
| ITS | = | internal transcribed spacer |
| LSU | = | large subunit rRNA |
| p.i. | = | post infection |
Acknowledgments
The authors are most thankful to Dr. Richard Bou Aoun and Marie-Céline Lafarge for clearing the initial microsporidia infestation, Dr. Laurent Troxler for generous help in the phylogenetic analysis and Dr. Caspar Franzen for kindly providing the MRC-5 cell line and T. ratisbonensis spores. S. N. has been supported by the Simone and Cino Del Duca Foundation (young team award to D. F.) and by the Collège Doctoral Européen (Strasbourg, France). D. F. is supported by the Centre National de la Recherche Scientifique and the team is an “Equipe Foundation Recherche Médicale.”
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Notes
† Current affiliation: UMR1225 IHAP; Ecole Nationale Vétérinaire de Toulouse; Toulouse Cedex, France
‡ Current affiliation: Max-Planck Institute of Immunobiology and Epigenetics; Freiburg, Germany
§ Current affiliation: Department of Medicine; Radboud University Nijmegen Medical Centre; Nijmegen, The Netherlands
||Current affiliation: Nijmegen Institute for Infection; Inflammation and Immunity (N4i); Radboud University Nijmegen Medical Centre; Nijmegen, The Netherlands
* Correspondence to: Dominique Ferrandon; Email: [email protected]
References
- Lemaitre B, Hoffmann J. The host defense of Drosophila melanogaster. Annu Rev Immunol 2007; 25:697 - 743; http://dx.doi.org/10.1146/annurev.immunol.25.022106.141615; PMID: 17201680
- Ferrandon D, Imler JL, Hetru C, Hoffmann JA. The Drosophila systemic immune response: sensing and signalling during bacterial and fungal infections. Nat Rev Immunol 2007; 7:862 - 74; http://dx.doi.org/10.1038/nri2194; PMID: 17948019
- Ashburner M, Golic GK, Hawley RS. Drosophila. A laboratory handbook. Cold Spring Harbor: Cold Spring Harbor Laboratory Press, 2004.
- Katinka MD, Duprat S, Cornillot E, Méténier G, Thomarat F, Prensier G, et al. Genome sequence and gene compaction of the eukaryote parasite Encephalitozoon cuniculi. Nature 2001; 414:450 - 3; http://dx.doi.org/10.1038/35106579; PMID: 11719806
- Franzen C. Microsporidia: how can they invade other cells?. Trends Parasitol 2004; 20:275 - 9; http://dx.doi.org/10.1016/j.pt.2004.04.009; PMID: 15147678
- Texier C, Vidau C, Viguès B, El Alaoui H, Delbac F. Microsporidia: a model for minimal parasite-host interactions. Curr Opin Microbiol 2010; 13:443 - 9; http://dx.doi.org/10.1016/j.mib.2010.05.005; PMID: 20542726
- Vizoso DB, Ebert D. Phenotypic plasticity of host-parasite interactions in response to the route of infection. J Evol Biol 2005; 18:911 - 21; http://dx.doi.org/10.1111/j.1420-9101.2005.00920.x; PMID: 16033563
- Armstrong E. Transmission and infectivity studies on Nosema kingi in Drosophila willistoni and other Drosophilids. Z Parasitenkd 1976; 50:161 - 5; http://dx.doi.org/10.1007/BF00380520
- Armstrong E. Transmission of Nosema kingi to offspring of Drosophila willistoni during copulation. Z Parasitenkd 1977; 53:311 - 5; http://dx.doi.org/10.1007/BF00389949; PMID: 413272
- Futerman PH, Layen SJ, Kotzen ML, Franzen C, Kraaijeveld AR, Godfray HC. Fitness effects and transmission routes of a microsporidian parasite infecting Drosophila and its parasitoids. Parasitology 2006; 132:479 - 92; http://dx.doi.org/10.1017/S0031182005009339; PMID: 16318674
- Vijendravarma RK, Godfray HC, Kraaijeveld AR. Infection of Drosophila melanogaster by Tubulinosema kingi: stage-specific susceptibility and within-host proliferation. J Invertebr Pathol 2008; 99:239 - 41; http://dx.doi.org/10.1016/j.jip.2008.02.014; PMID: 18394642
- Shaw RW, Kent ML. Fish microsporidia. In: Wittner M, ed. The microsporidia and microsporidiosis. Washington, D. C.: American society for microbiology, 1999:418-46.
- Didier ES, Weiss LM. Microsporidiosis: not just in AIDS patients. Curr Opin Infect Dis 2011; 24:490 - 5; http://dx.doi.org/10.1097/QCO.0b013e32834aa152; PMID: 21844802
- Troemel ER. New models of microsporidiosis: infections in Zebrafish, C. elegans, and honey bee. PLoS Pathog 2011; 7:e1001243; http://dx.doi.org/10.1371/journal.ppat.1001243; PMID: 21379567
- Franzen C, Fischer S, Schroeder J, Schölmerich J, Schneuwly S. Morphological and molecular investigations of Tubulinosema ratisbonensis gen. nov., sp. nov. (Microsporidia: Tubulinosematidae fam. nov.), a parasite infecting a laboratory colony of Drosophila melanogaster (Diptera: Drosophilidae). J Eukaryot Microbiol 2005; 52:141 - 52; http://dx.doi.org/10.1111/j.1550-7408.2005.04-3324.x; PMID: 15817119
- Kramer JP. Nosema kingi sp. n., a microsporidian from Drosophila willistoni Sturtevant, and its infectivity for other muscoides. J Insect Pathol 1964:491-9.
- Burnett RG, King RC. Observations on a microsporidian parasite of Drosophila willistoni Sturtevant. Journal of Insect Pathology 1962:104-12.
- Stalker HD, Carson HL. A very serious parasite of laboratory Drosophila. Drosophila Inform Serv 1963:96.
- Chatton E, Krempf A. Sur le cycle évolutif et la position systématique des protistes du genre Octosporea Flu, parasites des muscides. Bull Soc Zoll Fr 1911:172-9.
- Sin N, Meng L, Wang MQ, Wen JJ, Bornmann WG, Crews CM. The anti-angiogenic agent fumagillin covalently binds and inhibits the methionine aminopeptidase, MetAP-2. Proc Natl Acad Sci U S A 1997; 94:6099 - 103; http://dx.doi.org/10.1073/pnas.94.12.6099; PMID: 9177176
- Ferguson JA, Watral V, Schwindt AR, Kent ML. Spores of two fish microsporidia (Pseudoloma neurophilia and Glugea anomala) are highly resistant to chlorine. Dis Aquat Organ 2007; 76:205 - 14; http://dx.doi.org/10.3354/dao076205; PMID: 17803106
- Fallon JP, Reeves EP, Kavanagh K. The Aspergillus fumigatus toxin fumagillin suppresses the immune response of Galleria mellonella larvae by inhibiting the action of haemocytes. Microbiology 2011; 157:1481 - 8; http://dx.doi.org/10.1099/mic.0.043786-0; PMID: 21349977
- Franzen C, Fischer S, Schroeder J, Bleiss W, Schneuwly S, Schölmerich J, et al. In vitro cultivation of an insect Microsporidian Tubulinosema ratisbonensis in mammalian cells. J Eukaryot Microbiol 2005; 52:349 - 55; http://dx.doi.org/10.1111/j.1550-7408.2005.00043x; PMID: 16014013
- Vossbrinck CR, Baker MD, Didier ES, Debrunner-Vossbrinck BA, Shadduck JA. Ribosomal DNA sequences of Encephalitozoon hellem and Encephalitozoon cuniculi: species identification and phylogenetic construction. J Eukaryot Microbiol 1993; 40:354 - 62; http://dx.doi.org/10.1111/j.1550-7408.1993.tb04928.x; PMID: 8389641
- Corpet F. Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res 1988; 16:10881 - 90; http://dx.doi.org/10.1093/nar/16.22.10881; PMID: 2849754
- Rzhetsky A, Nei M. A simple method for estimating and testing minimum evolution trees. Mol Biol Evol 1992; •••:945 - 67
- Felsenstein J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985; •••:783 - 91; http://dx.doi.org/10.2307/2408678
- Tamura K, Nei M, Kumar S. Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc Natl Acad Sci U S A 2004; 101:11030 - 5; http://dx.doi.org/10.1073/pnas.0404206101; PMID: 15258291
- Nei M, Kumar S. Molecular Evolution and Phylogenetics. New York: Oxford University Press, 2000.
- Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 1987; 4:406 - 25; PMID: 3447015
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 2011; 28:2731 - 9; http://dx.doi.org/10.1093/molbev/msr121; PMID: 21546353
- Jung A, Criqui M-C, Rutschmann S, Hoffmann J-A, Ferrandon D. A microfluorometer assay to measure the expression of β-galactosidase and GFP reporter genes in single Drosophila flies. Biotechniques 2001; 30:594 - 601; PMID: 11252795