
Abstract
Traditionally, mouse humanization studies have used human fecal transfer to germ-free animals. This practice requires gnotobiotic facilities and is restricted to gnotobiotic mouse lines, which limits humanized mouse research. We have developed a generalizable method to humanize non germ-free mice using antibiotic treatment and human fecal transfer. The method involves depleting resident intestinal microbiota with broad-spectrum antibiotics, introducing human microbiota from frozen fecal samples by weekly gavage, and maintaining mice in HEPA-filtered microisolator cages. Pyrosequencing cecal microbiota 16S rRNA genes showed that recipient mice adopt a humanized microbiota profile analogous to their human donors, and distinct from mice treated with only antibiotics (no fecal transfer) or untreated control mice. In the humanized mice, 75% of the sequence mass was observed in their respective human donor and conversely, 68% of the donor sequence mass was recovered in the recipient mice. Principal component analyses of GC- and HPLC-separated cecal metabolites were performed to determine effects of transplanted microbiota on the metabolome. Cecal metabolite profiles of mice treated with only antibiotics (no fecal transfer) and control mice were dissimilar from each other and from humanized mice. Metabolite profiles for mice humanized from different donor samples clustered near each other, yet were sufficiently distinct that separate clusters were apparent for each donor. Also, cecal concentrations of 57 metabolites were significantly different between humanization treatments. These data demonstrate that our protocol can be used to humanize non germ-free mice and is sufficiently robust to generate metabolomic differences between mice humanized from different human donors.
Introduction
The lumen of the colon is home to an estimated 100 trillion bacteriaCitation1 with a gene set approximately 150-fold larger than the human genome.Citation2 Recent evidence suggests that the gut microbiota composition and associated metabolic capabilities can have far-reaching effects on systemic metabolism, physiology and overall health. Gut microbiota are now recognized as key factors in the etiology of several important human diseases, including obesity,Citation3,Citation4 metabolic syndrome,Citation5,Citation6 inflammation,Citation5,Citation7 and colorectal cancer.Citation8,Citation9 The field of gut microbiology and associated health outcomes has advanced greatly through the use of “humanized” mouse models.Citation10-Citation12 Traditionally, these models require seeding germ-free mice with bacteria derived from human donors, thus providing an useful system to study the interactions between human microbiota and chronic disease in studies where human subjects are not appropriate. However, maintenance of germ-free mice colonies is expensive, requiring substantial institutional investment in infrastructure and specialized personnel. Moreover, germ-free mice are not readily available for the most common and/or most important strains used in chronic disease research, including many inbred and genetic mouse models.
To overcome these limitations, we have developed a simplified method to replace microbiota normally present in rodents with microbiota harvested from human fecal samples. Briefly, the method involves: (1) depleting resident animal intestinal microbiota by gavaging the animal twice daily for 17 d with broad spectrum antibiotics (ampicillin, vancomycin, neomycin, and metronidazole) and an antifungal (amphotericin B); (2) introducing human microbiota, derived from frozen fecal samples, to the mouse gastrointestinal tract by weekly oral gavage; and (3) maintaining the animals in microisolator cages supplied with HEPA filtered air.
Results
Phenotypic differences
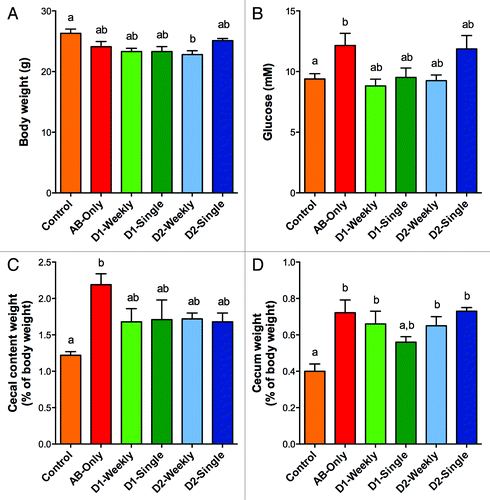
Compared with controls, mice that received the antibiotic depletion cocktail had the same final body weight regardless of microbiota repletion, except for mice gavaged weekly with microbiota from Donor 2 (P < 0.05, ). Interestingly, only mice treated with antibiotics, but not dosed with microbiota, had higher fasting glucose levels than controls (P < 0.05, ). Cecal content weight was greater in antibiotic-only treated mice relative to controls (P < 0.05, ). Similarly, cecal weight was significantly greater in all mice that received antibiotic treatment compared with control mice except for mice inoculated singly with microbiota from Donor 1 (P < 0.05, ).
Figure 1. Effect of antibiotic treatment and fecal transfer with human microbiota on (A) final body weight, (B) fasting, whole blood glucose, (C) cecal content weight, and (D) empty cecum weight. Experimental groups are as follows: Control, no antibiotic treatment or fecal transplant; AB-Only, mice dosed with antibiotics but no fecal transfer; D1-Weekly, mice gavaged weekly with microbiota from donor 1; D1-Single, mice gavaged once with microbiota from donor 1; D2-Weekly, mice gavaged weekly with microbiota from donor 2; D2-Single, mice gavaged once with microbiota from donor 2. Values shown are average ± SD (n = 4 to 10 mice). Groups indicated with different labels are significantly different (P < 0.05) as determined by one-way ANOVA with Tukey’s post-hoc test for multiple comparisons.
Microbiota profiles
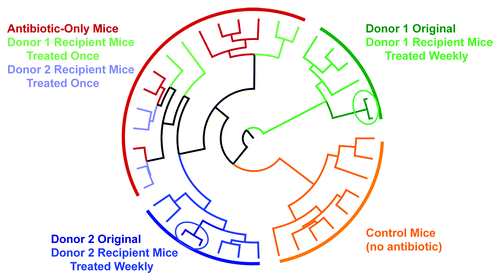
Evidence from microbiota profiling shows that the recipient animals adopted distinct intestinal microbiota profiles reflective of their human donors. Weighted Unifrac analysis of the cecal microbiota population differentiated control mice (no antibiotics), antibiotic-only treated mice, and mice receiving either Donor 1 or 2 inoculant on a weekly basis into separate clades (). Importantly, the microbiota populations in donor fecal samples and their weekly-dosed recipient mice cecal samples co-segregate into the same distinct clades. Mice dosed only once with bacteria from either donor co-segregated into the same clade as antibiotic-only treated mice. This observation suggests that weekly inoculations are necessary for successful and sustained establishment of the donor microbiome in the recipient animal using this antibiotic-based approach ().
Figure 2. Weighted Unifrac tree of cecal microbiota. Branches were colored according to the sample material as follows: orange, control mice no antibiotics or fecal transfer; red, antibiotic-only mice, no fecal transfer; dark green, Donor 1 original source bacteria; light green, mice dosed singly with bacteria from Donor 1; medium green, mice dosed weekly with bacteria from donor 1; dark blue, Donor 2 original source bacteria; light blue, mice dosed singly with bacteria from Donor 2; medium blue, mice dosed weekly with bacteria from Donor 2.
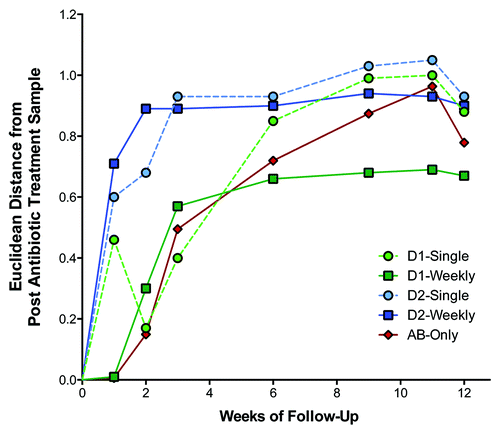
To characterize the time course of microbiota colonization after antibiotic treatment, TRFLP analysis was performed using mouse fecal bacteria samples that were collected over 12 wk. Results of this analysis showed that microbial populations in weekly gavaged recipients tended to stabilize after approximately 3 wk (). In the antibiotic treated mice not dosed with human bacteria, the microbiota population remained stable for one week, but quickly changed thereafter. Results for mice receiving a single gavage of human donor bacteria were variable ().
Figure 3. Time-course of changes in fecal microbiota. Data are based on terminal restriction fragment length polymorphism analysis of fecal bacterial collected at each time point. Values shown are the mean Euclidean distance from the immediate post antibiotic treatment sample derived from principle coordinate vectors 1, 2, and 3 of the Pearson population distance matrix. Abbreviations are D1, Donor 1; D2, Donor 2; and AB, antibiotic.
In mice dosed with human gut microbiota on a weekly basis, 76% (Donor 1) and 66% (Donor 2) of the mouse sequence mass (e.g., percent of the total number of sequences) was reflected in the corresponding human donor sample. In contrast, in control mice 6% (Donor 1) and 37% (Donor 2) of the sequence masses were also found in human donor samples. The relatively higher congruence between control mice and Donor 2 was the result of two prokMSA IDs that were coincidentally abundant in the control mice (combined, 27% of the sequence mass) and also present in Donor 2 but at much lower levels (combined, 0.06% of the sequence mass, and not present in Donor 1.
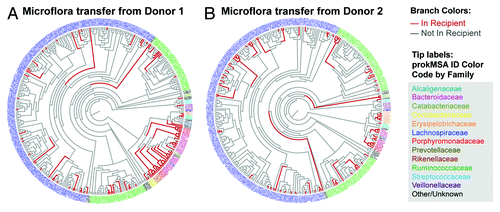
A taxonomic analysis of the bacterial lineages revealed that mice that were dosed with microbiota from Donor 1 acquired a maximum of 28% of the donor prokMSA IDs representing 57% of the donor sequence mass. Similarly, mice dosed with bacteria from Donor 2 acquired up to 27% of prokMSA IDs, representing 68% of the donor sequence mass (). This taxonomic analysis also indicated that transfer of bacteria from donor to recipient was particularly efficient for Bacteriodetes families, including Bacteroidaceae, Rikenellaceae, Porphyromonadaceae, and the Coriobacteriaceae family from the Actinobacteria phylum (). In contrast, colonization of bacteria from the Lachnospiraceae and Ruminococcaceae families of the Firmicute phylum was apparently inefficient.
Figure 4. Bacterial lineages in (A) Donor 1 found in Donor 1 recipient mice gavaged weekly and (B) Donor 2 found in Donor 2 recipient mice gavaged weekly. prokMSA IDs are colored according to bacteria families, as indicated in the figure legend, while each branch is colored according to its detection in the recipient mouse, either not detected (gray) or positively detected (red).
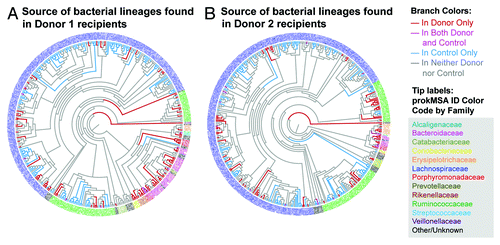
Next, taxonomic analyses of mouse recipient cecal bacterial family lineages were performed to determine whether the lineage was common exclusively with the human donor, common exclusively with the recipient mouse, or shared with both the human donor and the control mice (). Fifty-seven and 39% of the sequence mass in mice receiving bacteria from Donors 1 and 2, respectively, were exclusively in common with the corresponding human donor, while 19 and 27% of sequence mass was in common with both the human donor and control mice respectively. Only 22 and 31% of sequence mass in mice receiving bacteria from Donors 1 and 2, respectively, were exclusively in common with the control mice, while 2 and 3% of sequence mass were not in common with either the human donor or control mice ().
Figure 5. Source of bacterial lineages found in (A) Donor 1 recipient mice gavaged weekly and (B) Donor 2 recipient mice gavaged weekly. prokMSA IDs are colored according to bacteria families, as indicated in the figure legend, while each branch is colored according to its source, as follows: red, detected in the donor only; pink, detected in both the donor and recipient mouse; blue, detected in control mice only; and gray, detected in neither control or donor.
Metabolomic profiles
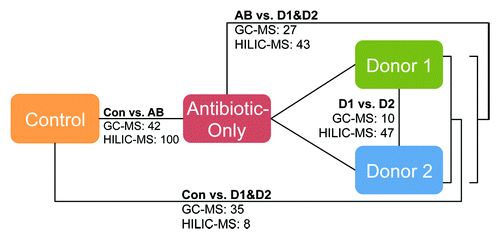
A metabolomic analysis of cecal metabolites was performed to determine whether the fecal transfer method employed in this study impacted microbial metabolism. Metabolites for mice in the control, antibiotic-only, Donor 1 weekly and Donor 2 weekly groups were measured by GC-MS and hydrophilic interaction liquid chromatography (HILIC)-MS. Partial least squares discriminate analyses revealed that metabolic profiles for animals clustered according to treatment groups; control and antibiotic-only mice segregated into clusters distinct from mice dosed with human microbiota (). Additionally, metabolic profiles for each of the two human donors were also distinct, most notably for metabolites detected by HILIC-MS (). highlights differences in metabolites detected by GC-MS or HILIC-MS among groups, and Tables S1–S8 provide lists of specific metabolites and results of pair-wise comparisons between each of the test groups. Antibiotic treatment strongly altered the cecal metabolome as indicated by the highest number of significantly altered metabolites compared with untreated control mice. The metabolite profiles for antibiotic-only and control mice are substantially dissimilar from each other and from the metabolite profiles for the humanized mice. Profile comparison revealed that 142 and 70 cecal metabolites (GC-MS and HILIC-MS, respectively) were distinct between mice treated with antibiotic-only and either the control or humanized mice. Collectively, the humanized mice had a more similar cecal metabolome to the control mice than the antibiotic-only mice. Of particular note, a total of 57 cecal metabolites were present at significantly different levels in mice humanized with bacteria from Donor 1 compared with Donor 2 (; Tables S1 and S2). This suggests our method can induce cecal metabolite differences dependent on the source of human microbiota. Because mice were fed identical chow diets, unique microbial populations resulting from our humanization protocol and the subsequent differential microbial metabolism likely explains these differences.
Figure 6. Metabolomic profile of cecal metabolites by partial least square discriminate analysis (PLS-DA) of (A) GC-MS and (B) HILIC-MS analytes. Abbreviations are as follows: Con, control mice (no antibiotic treatment or fecal transfer); AB, antibiotic treatment only; D1, mice dosed weekly with bacteria from Donor 1; and D2, mice dosed weekly with bacteria from Donor 2.
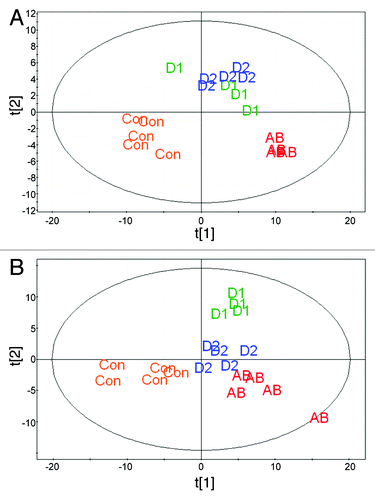
Figure 7. Effect of experimental treatments on the cecal metabolome. The numbers shown in the figure denote the number of significant, differentially expressed metabolites between treatments determined by either GC-MS or HILIC-MS. Listings of these metabolites and pairwise comparisons among sample groups are provided in Tables S1–S8.
Discussion
Differences in host genetics, diet, and digestive tract physiology present significant challenges for establishing mouse models of the human gut microbiome. In the present study, we describe a method to humanize non germ-free mice with antibiotic treatment and human fecal transfer. Taxonomic analyses of gut microbiota and metabolite profiles in mice humanized with bacteria from two human donors indicate that this approach results in distinct bacteria profiles in recipient mice that largely reflect their corresponding donors. The other principle method for humanizing the mouse microbiome requires the use of germ-free mice, yet this approach does not result in a complete transfer of donor bacteria to the recipient mice.Citation10,Citation13 Similarly, the antibiotic method described here also yielded an imperfect transfer of either human donor microbiome. Transfer of the Bacteriodetes phylum from the human donors to recipient mice was generally more successful compared with the Firmicutes phylum (). This is in accordance with previous studies using human fecal transfer to humanize germ free mice. Investigations by Turnbaugh et al.,Citation10 and Wos-Oxley et al.,Citation13 have both reported selective enrichment of Bacteroidetes compared with the Firmicute phylum after humanization of germ free mice.
Despite the imperfect transfer, data obtained in this study show that our fecal transfer protocol resulted in substantial changes to the cecal metabolome and that this method is sufficiently robust such that cecal metabolite differences between mice humanized with microbiota from different human donors were readily apparent. Microbiota inoculation from different human donors resulted in significantly different levels of 57 cecal metabolites in the recipient mice () despite being fed identical diets. This strongly suggests that the donor microbiota was the primary variable driving changes to the cecal metabolome. Therefore, this protocol could be used to humanize mice from well-established disease models for investigations into the contribution of human intestinal bacteria on the etiology of disorders linked to gut microbiota and microbial metabolites such as colon cancer, inflammatory bowel disease, obesity, diabetes and autism.Citation14-Citation22
Other groups have used antibiotics and fecal transfer to humanize rodents. Manichanh, et al. transplanted rat cecal material to rats treated for 3 d with a combination of vancomycin and imipenem.Citation23 They reported that a single gavage of cecal material following antibiotic treatment increased bacterial diversity compared with rats treated only with antibiotics, but this procedure did not shift the population of cecal microbiota so that it was more similar to the input microbiota population. In an experiment that compared humanization protocols, Wos-Oxley, et al. reported that C57BL/6 mice treated with antibiotics and inoculated with human fecal material harbored a microbiome that was more similar to untreated, control mice than germ free cohorts humanized with the same donor feces.Citation13 In the Wos-Oxley study, mice in the antibiotic humanization treatment were provided ciprofloxacin orally via the drinking water for four days and gavaged with donor feces for five consecutive days. In the current study, it was determined that a single gavage of donor sample was insufficient to humanize mice after antibiotic treatment, whereas repeated weekly gavages were necessary to emulate the human donor microbiota population.
The antibiotic protocol employed in this study was more rigorous than was used in these prior studies. Mice were gavaged every 12 h with a cocktail of four antibiotics for 14 consecutive days compared with the antibiotic depletion protocols used by Manichanh, et al. (two antibiotics for three days) or Wos-Oxley, et al. (one antibiotic for four days).Citation13,Citation23 These differences in protocols may be critical in establishing a humanized microbiome in antibiotic treated animals. In our study, mice that were inoculated weekly with donor feces had a microbiome that was more similar to their respective human donors than control mice, while mice inoculated singly with donor feces were most similar to mice only treated with antibiotics (). However, it is currently not clear how many weekly gavages are necessary to maintain the humanized profile. Weekly inoculations may only be necessary for only the initial two to three weeks post antibiotic treatment (). Thus, future work is needed to determine the optimum inoculation protocol.
Although the approach to humanize the gut microbiome of laboratory animals outlined in this report is technologically straightforward, this method clearly has potential to impact this field of science. Other investigators who have successfully humanized mouse intestinal microbiota relied on the use of germ-free mice as recipients and subsequent maintenance of the animals in a dedicated, germ-free vivarium.Citation10 However, this approach has substantial (and potentially insurmountable) limitations for many investigators. Most important of these is the availability of germ-free mice strains. The vast majority of well-characterized inbred mouse strains and genetically modified mice, many of which are essential models for the study of human disease, are not commercially available in germ-free form. This resource limitation represents a significant obstacle for the vast majority of research groups who wish to examine the impact of human microbiota populations in established animal models of human disease, but lack the means to derive germ-free animals from the appropriate strain of interest. Mice of any strain or genetic model can have their intestinal microbiota humanized on demand as needed by the investigator following the methods outlined in this paper. Using our approach, mice maintained a humanized profile for 12 wk. Mouse models of human chronic diseases that are influenced by gut microbiota such as obesityCitation4,Citation6,Citation24,Citation25 and colon cancerCitation8,Citation9,Citation21,Citation26,Citation27 typically require prolonged studies lasting several months.Citation28,Citation29 Also of note, this new approach can likely be extended to other frequently used animal model species such as rats and hamsters.
Materials and Methods
Microbiota depletion
Seven week old, male, A/J strain mice were randomized into the following treatment groups: control (no antibiotic treatment, no fecal transfer, n = 10); Donor 1 humanized, one-time fecal transfer (n = 4); Donor 1 humanized, weekly fecal transfer (n = 4); Donor 2 humanized, one-time fecal transfer (n = 4); Donor 2 humanized, weekly fecal transfer (n = 5); and antibiotic treated, no fecal transfer (n = 7). Except for the control treatment, mice were gavaged every 12 h with 1 mg/kg amphotericin-B for 3 d followed by an antibiotic cocktail described Reikvam, et al.Citation30 consisting of 50 mg/kg vancomycin, 100 mg/kg neomycin, 100 mg/kg metronidazole, and 1 mg/kg amphotericin-B for 14 d. Ampicillin was provided in drinking water (1 g/L) ad libitum. All mice were maintained in autoclaved microisolator cages supplied with HEPA-filtered air, fed irradiated chow (Harlan) and provided with autoclaved water. Freshly voided fecal samples were collected weekly.
Humanization protocol
Twelve hr after the final antibiotic gavage, mice in the fecal transfer treatments were gavaged with fecal material reconstituted in sterile saline (1 g/10 mL) from one of two human donors (Donor 1 or 2). Mice in the weekly fecal transfer treatments were gavaged weekly for 12 wk with fecal material from their respective donors. At 12 wk after the antibiotic depletion protocol, all mice were euthanized after a four-hour fasting period by CO2 asphyxiation and cardiac venipuncture. Plasma glucose concentrations were measured using a commercial kit (Sigma-Aldrich, Co), and cecal contents were snap-frozen in liquid nitrogen. All animal care and experimental procedures were approved by the Utah State University Institutional Animal Care and Use Committee.
Human fecal collection and processing
The human donors were provided with disposable coffee filter-like fecal collection devices, and samples were frozen at -80 °C immediately after collection. The QIAamp DNA stool mini kit (Qiagen) was used to extract fecal bacterial DNA as described by the manufacturer, with the exceptions that bead beating was incorporated into the protocol to increase bacterial lysis and the temperature of the extraction was increased to 95 °C.
Estimation of microbiota population stability
Terminal restriction fragment length polymorphism (TRFLP)Citation31 analysis was employed to estimate changes in microbiota population structure over time. Briefly, fecal DNA samples collected at each time point were amplified using the primers 8–27f: 5'6-FAM-AGA GTT TGA TCM TGG CTC AG-3' and 1512–1492r: 5'-HEX-ACG GYT ACC TTG TTA CGA CTT-3'. PCR products were digested with HaeIII. Internal standards comprised of HAEIII digested PCR products of Enterococcus faecalis, Ruminococcus gnavus, Bifidobacterium breve, Escherichia coli, and Bacteroides thetaiotaomicron labeled on the 5' end with NED were added to each sample prior to sizing of the restriction fragments (ABI PRISM 3730 DNA Analyzer).
Restriction fragment sizes (both 5' and 3') were adjusted for slight lane-to-lane and run-to-run variations based on the apparent size of the internal standards. Identified restriction fragments from all samples were combined into a single file, sorted on size and initially binned based on identified “breaks” (distance between adjacent restriction fragments > 0.5 bp) in the size distribution. Bins covering a size range of >1.2 bp or containing 2 or more restriction fragments from the sample were further subdivided based on narrower breaks in the size distribution of the restriction fragments until all bins spanned a size range of ≤1.2 bp and contained no more than one restriction fragment from a given sample. Each restriction fragment was subsequently assigned a size equal to the median of all restriction fragments within their assigned bin.
For both 5' and 3' ends, restriction fragment sizes were included in the statistical analysis if they appeared in the top 10 most abundant restriction fragments in any sample. Selected 5' and 3' restriction fragments were combined into a single data set and subjected to principal component analysis. Change in microbiota population composition was summarized as the Euclidean distance from week 0 of each sample based on the first three principal component vectors.
Microbiota characterization
Cecal contents were collected after euthanasia, weighed and snap frozen in liquid N2. Isolated cecal DNA was amplified using barcoded primers directed against the V1+V2 region of the 16S rRNA.Citation25,Citation32 Equal amounts of the PCR products with different barcodes were combined, amplified and sequenced on a PicoTiterPlate with a Roche 454 GS FLX Titanium System (Roche 454 Life Sciences) using protocols adapted by the Center for Integrated BioSystems at Utah State University. Microbiota sequences were processed through the latest version (1.6) of QIIME pyrosequencing data analysis pipeline.Citation33 Following data quality filtering and assignment of the barcoded multiplex reads to their appropriate samples, the sequences were filtered, chimera-checked and clustered into operational taxonomic units (OTUs) at a 97% sequence similarity using OTUPipeCitation34,Citation35 Taxonomic assignments for representative sequences were made with RDP ClassifierCitation36 using a confidence level of 0.50 and employing the Greengenes reference OUT database (release 12_10).Citation37 OTUs with identical assigned prokMSA were combined for subsequent analyses. The sequences were aligned with PyNASTCitation38 and a phylogenetic tree constructed with FastTreeCitation39 after the aligned sequences were filtered with the default lanemask file. Weighted UniFrac analysisCitation40 was used to estimate the phylogenetic distances between donors and recipient microbial populations.
Metabolomics sample preparation
The cecal material was extracted using a modified hot ethanol procedure. Five mL of boiling 75% EtOH (v/v) was added to each fecal sample, vortexed and then incubated at 90 °C for 5 min. Cell debris was removed by centrifugation at 5000 × g for 5 min. The supernatant was equally divided into two new tubes and dried under a vacuum.
GC-MS analysis of the metabolome
All GC-MS analysis was performed with a Waters GCT Premier mass spectrometer (Waters Corp) fitted with an Agilent 6890 gas chromatograph (Agilent Technologies). Dried samples were suspended in 40 µL of 40 mg/mL O-methoxylamine hydrochloride in pyridine and incubated for one hour at 30 °C. Twenty-five µL of this solution with 20 µL of N-Methyl-N-(trimethylsilyl)trifluoroacetamide was added to each autosampler vial and incubated for 30 min at 37 °C with shaking. Next, 1 µL of sample was injected into the 250 °C inlet at a 10:1 split. Gas chromatography with an initial temperature of 95 °C for 1 min followed by a 40 °C per min increase in temperature to 110 °C and a hold time of 2 min, then a second period of temperature increase of 5 °C per min to 250 °C, and finally a third ramp to 350 °C with a final hold time of 3 min. A 30 min Phenomenex ZB-5MSi column and a 5 min guard column were employed for gas chromatography analysis. Data were collected using MassLynxTM mass spectrometry software, version 4.1 (Waters Corp), while peak identification was performed using MarkerLynxTMXS software (Waters Corp).
LC-MS analysis of the metabolome
All LC-MS analysis was performed using an Agilent 6520 QTOF fitted with an Agilent 1100 HPLC (Agilent Technologies). For separation, a 100 × 2.1 mm, 3.5 µm, 100A SeQuant zic-HILIC column (Merck Millipore) was used. Ammonium formate pH 3.2 was used as the strong solvent, with acetonitrile the weak solvent. The initial LC condition was held at 90% acetonitrile for one minute followed by a 20 min ramp to 40% acetonitrile and a 2 min hold. The column was re-equilibrated over 10 min at 90% acetonitrile. Flow rate was 0.2 mL/min. Data were recorded and translated to mzdata file format using MassHunter Workstation software (Agilent Technologies), and then processed using MZmine software, version 2.1.Citation41
Additional material
Download Zip (2.1 MB)Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
Acknowledgments
The authors wish to thank Dr Aaron Olsen for technical assistance as well as Brett Healy, Deanna Larson, and Nancy Hergert for assisting with experiments. Funding was provided by the Utah Science Technology and Research Initiative and the Utah Agricultural Experiment Station.
References
- Berg RD. The indigenous gastrointestinal microflora. Trends Microbiol 1996; 4:430 - 5; http://dx.doi.org/10.1016/0966-842X(96)10057-3; PMID: 8950812
- Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, et al, MetaHIT Consortium. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010; 464:59 - 65; http://dx.doi.org/10.1038/nature08821; PMID: 20203603
- Turnbaugh PJ, Gordon JI. The core gut microbiome, energy balance and obesity. J Physiol 2009; 587:4153 - 8; http://dx.doi.org/10.1113/jphysiol.2009.174136; PMID: 19491241
- Delzenne NM, Neyrinck AM, Bäckhed F, Cani PD. Targeting gut microbiota in obesity: effects of prebiotics and probiotics. Nat Rev Endocrinol 2011; 7:639 - 46; http://dx.doi.org/10.1038/nrendo.2011.126; PMID: 21826100
- Kau AL, Ahern PP, Griffin NW, Goodman AL, Gordon JI. Human nutrition, the gut microbiome and the immune system. Nature 2011; 474:327 - 36; http://dx.doi.org/10.1038/nature10213; PMID: 21677749
- Cani PD, Osto M, Geurts L, Everard A. Involvement of gut microbiota in the development of low-grade inflammation and type 2 diabetes associated with obesity. Gut Microbes 2012; 3:279 - 88; http://dx.doi.org/10.4161/gmic.19625; PMID: 22572877
- Garrett WS, Gordon JI, Glimcher LH. Homeostasis and inflammation in the intestine. Cell 2010; 140:859 - 70; http://dx.doi.org/10.1016/j.cell.2010.01.023; PMID: 20303876
- Arthur JC, Jobin C. The complex interplay between inflammation, the microbiota and colorectal cancer. Gut Microbes 2013; 4:253 - 8; http://dx.doi.org/10.4161/gmic.24220; PMID: 23549517
- Macdonald RS, Wagner K. Influence of dietary phytochemicals and microbiota on colon cancer risk. J Agric Food Chem 2012; 60:6728 - 35; http://dx.doi.org/10.1021/jf204230r; PMID: 22632581
- Turnbaugh PJ, Ridaura VK, Faith JJ, Rey FE, Knight R, Gordon JI. The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Sci Transl Med 2009; 1:ra14; http://dx.doi.org/10.1126/scitranslmed.3000322; PMID: 20368178
- Goodman AL, Kallstrom G, Faith JJ, Reyes A, Moore A, Dantas G, Gordon JI. Extensive personal human gut microbiota culture collections characterized and manipulated in gnotobiotic mice. Proc Natl Acad Sci U S A 2011; 108:6252 - 7; http://dx.doi.org/10.1073/pnas.1102938108; PMID: 21436049
- Gootenberg DB, Turnbaugh PJ. Companion animals symposium: humanized animal models of the microbiome. J Anim Sci 2011; 89:1531 - 7; http://dx.doi.org/10.2527/jas.2010-3371; PMID: 20833767
- Wos-Oxley M, Bleich A, Oxley AP, Kahl S, Janus LM, Smoczek A, Nahrstedt H, Pils MC, Taudien S, Platzer M, et al. Comparative evaluation of establishing a human gut microbial community within rodent models. Gut Microbes 2012; 3:234 - 49; http://dx.doi.org/10.4161/gmic.19934; PMID: 22572831
- Tehrani AB, Nezami BG, Gewirtz A, Srinivasan S. Obesity and its associated disease: a role for microbiota?. Neurogastroenterol Motil 2012; 24:305 - 11; http://dx.doi.org/10.1111/j.1365-2982.2012.01895.x; PMID: 22339979
- Kootte RS, Vrieze A, Holleman F, Dallinga-Thie GM, Zoetendal EG, de Vos WM, Groen AK, Hoekstra JB, Stroes ES, Nieuwdorp M. The therapeutic potential of manipulating gut microbiota in obesity and type 2 diabetes mellitus. Diabetes Obes Metab 2012; 14:112 - 20; http://dx.doi.org/10.1111/j.1463-1326.2011.01483.x; PMID: 21812894
- Musso G, Gambino R, Cassader M. Interactions between gut microbiota and host metabolism predisposing to obesity and diabetes. Annu Rev Med 2011; 62:361 - 80; http://dx.doi.org/10.1146/annurev-med-012510-175505; PMID: 21226616
- Cucchiara S, Stronati L, Aloi M. Interactions between intestinal microbiota and innate immune system in pediatric inflammatory bowel disease. J Clin Gastroenterol 2012; 46:Suppl S64 - 6; http://dx.doi.org/10.1097/MCG.0b013e31826a857f; PMID: 22955361
- Lawrance IC. Microbiota and management of inflammatory bowel disease. J Gastroenterol Hepatol 2012; 27:1137 - 40; http://dx.doi.org/10.1111/j.1440-1746.2012.07163.x; PMID: 22712707
- Kinross JM, von Roon AC, Holmes E, Darzi A, Nicholson JK. The human gut microbiome: implications for future health care. Curr Gastroenterol Rep 2008; 10:396 - 403; http://dx.doi.org/10.1007/s11894-008-0075-y; PMID: 18627653
- Marteau P, Chaput U. Bacteria as trigger for chronic gastrointestinal disorders. Dig Dis 2011; 29:166 - 71; http://dx.doi.org/10.1159/000323879; PMID: 21734380
- Rowland IR. The role of the gastrointestinal microbiota in colorectal cancer. Curr Pharm Des 2009; 15:1524 - 7; http://dx.doi.org/10.2174/138161209788168191; PMID: 19442169
- Iebba V, Aloi M, Civitelli F, Cucchiara S. Gut microbiota and pediatric disease. Dig Dis 2011; 29:531 - 9; http://dx.doi.org/10.1159/000332969; PMID: 22179208
- Manichanh C, Reeder J, Gibert P, Varela E, Llopis M, Antolin M, Guigo R, Knight R, Guarner F. Reshaping the gut microbiome with bacterial transplantation and antibiotic intake. Genome Res 2010; 20:1411 - 9; http://dx.doi.org/10.1101/gr.107987.110; PMID: 20736229
- Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL, Griffin NW, Lombard V, Henrissat B, Bain JR, et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 2013; 341:1241214; http://dx.doi.org/10.1126/science.1241214; PMID: 24009397
- Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, et al. A core gut microbiome in obese and lean twins. Nature 2009; 457:480 - 4; http://dx.doi.org/10.1038/nature07540; PMID: 19043404
- Marchesi JR, Dutilh BE, Hall N, Peters WH, Roelofs R, Boleij A, Tjalsma H. Towards the human colorectal cancer microbiome. PLoS One 2011; 6:e20447; http://dx.doi.org/10.1371/journal.pone.0020447; PMID: 21647227
- McCoy AN, Araújo-Pérez F, Azcárate-Peril A, Yeh JJ, Sandler RS, Keku TO. Fusobacterium is associated with colorectal adenomas. PLoS One 2013; 8:e53653; http://dx.doi.org/10.1371/journal.pone.0053653; PMID: 23335968
- Surwit RS, Feinglos MN, Rodin J, Sutherland A, Petro AE, Opara EC, Kuhn CM, Rebuffé-Scrive M. Differential effects of fat and sucrose on the development of obesity and diabetes in C57BL/6J and A/J mice. Metabolism 1995; 44:645 - 51; http://dx.doi.org/10.1016/0026-0495(95)90123-X; PMID: 7752914
- Tanaka T, Kohno H, Suzuki R, Yamada Y, Sugie S, Mori H. A novel inflammation-related mouse colon carcinogenesis model induced by azoxymethane and dextran sodium sulfate. Cancer Sci 2003; 94:965 - 73; http://dx.doi.org/10.1111/j.1349-7006.2003.tb01386.x; PMID: 14611673
- Reikvam DH, Erofeev A, Sandvik A, Grcic V, Jahnsen FL, Gaustad P, McCoy KD, Macpherson AJ, Meza-Zepeda LA, Johansen FE. Depletion of murine intestinal microbiota: effects on gut mucosa and epithelial gene expression. PLoS One 2011; 6:e17996; http://dx.doi.org/10.1371/journal.pone.0017996; PMID: 21445311
- Li F, Hullar MA, Lampe JW. Optimization of terminal restriction fragment polymorphism (TRFLP) analysis of human gut microbiota. J Microbiol Methods 2007; 68:303 - 11; http://dx.doi.org/10.1016/j.mimet.2006.09.006; PMID: 17069911
- Andersson AF, Lindberg M, Jakobsson H, Bäckhed F, Nyrén P, Engstrand L. Comparative analysis of human gut microbiota by barcoded pyrosequencing. PLoS One 2008; 3:e2836; http://dx.doi.org/10.1371/journal.pone.0002836; PMID: 18665274
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 2010; 7:335 - 6; http://dx.doi.org/10.1038/nmeth.f.303; PMID: 20383131
- Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010; 26:2460 - 1; http://dx.doi.org/10.1093/bioinformatics/btq461; PMID: 20709691
- Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011; 27:2194 - 200; http://dx.doi.org/10.1093/bioinformatics/btr381; PMID: 21700674
- Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 2007; 73:5261 - 7; http://dx.doi.org/10.1128/AEM.00062-07; PMID: 17586664
- DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 2006; 72:5069 - 72; http://dx.doi.org/10.1128/AEM.03006-05; PMID: 16820507
- Caporaso JG, Bittinger K, Bushman FD, DeSantis TZ, Andersen GL, Knight R. PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics 2010; 26:266 - 7; http://dx.doi.org/10.1093/bioinformatics/btp636; PMID: 19914921
- Price MN, Dehal PS, Arkin AP. FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol Biol Evol 2009; 26:1641 - 50; http://dx.doi.org/10.1093/molbev/msp077; PMID: 19377059
- Lozupone C, Hamady M, Knight R. UniFrac--an online tool for comparing microbial community diversity in a phylogenetic context. BMC Bioinformatics 2006; 7:371; http://dx.doi.org/10.1186/1471-2105-7-371; PMID: 16893466
- Pluskal T, Castillo S, Villar-Briones A, Oresic M. MZmine 2: modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinformatics 2010; 11:395; http://dx.doi.org/10.1186/1471-2105-11-395; PMID: 20650010