Abstract
Allergic drug reactions occur when a drug, usually a low molecular weight molecule, has the ability to stimulate an immune response. This can be done in one of two ways. The first is by binding covalently to a self-protein, to produce a haptenated molecule that can be processed and presented to the adaptive immune system to induce an immune response. Sometimes the drug itself cannot do this but a reactive breakdown product of the drug is able to bind covalently to the requisite self-protein or peptide. The second way in which drugs can stimulate an immune response is by binding non-covalently to antigen presenting or antigen recognition molecules such as the major histocompatibility complex (MHC) or the T cell receptor. This is known as the p-I or pharmacological interaction hypothesis. The drug binding in this situation is reversible and stimulation of the response may occur on first exposure, not requiring previous sensitization. There is probably a dependence on the presence of certain MHC alleles and T cell receptor structures for this type of reaction to occur.
There is increasing evidence for the need for specific MHC alleles to be present in order for drug reactions to occur. This requirement varies between ethnic groups and may be limited to certain drugs and certain forms of hypersensitivity reactions, such as Stevens-Johnson/Toxic epidermal necrolysis. It has also become more evident that there is an interaction between drug hypersensitivity reactions and viral infections, best known with the maculo-papular rashes occurring with amino-penicillins but clearly demonstrated in the drug-induced Hypersensitivity Syndrome, where an interplay of drug-induced immune responses and Herpes viruses occurs. There is increasing evidence for the ability of drugs to initiate immune responses through activation of the innate immune system. In addition, drug reactions can appear to involve the adaptive immune system when in fact the manifestations are due to direct effects upon mediator-containing cells such as mast cells, or other inflammatory systems such as prostaglandin/leukotrienes and the kinin system.
In the presence of drug hypersensitivity, it is sometimes necessary to re-institute administration of the implicated drug because no satisfactory alternatives are available. There have been significant advances in such techniques, particularly for reactions considered to be immediate or anaphylactic in type, and there is increased understanding of the mechanisms that may be involved in desensitization. However many drug reactions appear to involve cell-mediated immune responses, and while desensitization in milder forms of such drug hypersensitivity is performed, little is known of the mechanisms involved.
Adverse drug reactions are frequently classified into two types. Type A reactions are common and are caused by the pharmacologic or toxic effects of the drug. Type B reactions are uncommon and unpredictable, occurring in susceptible and predisposed individuals. These include allergic drug reactions, making up about 15% of all adverse drug reactions.1
Pathogenesis of Immune Response to Drugs
Because most drugs are low molecular weight chemicals, in theory too small to be able to stimulate the immune system, it has long been assumed that the drug or a reactive metabolite, must first bind covalently to a macromolecule such as a protein, forming a multivalent conjugate that is processed and presented by the immune system to T lymphocytes.Citation2 This mechanism would not be required for large molecular weight drugs that express multiple epitopes. These would include proteins or peptides used therapeutically such as monoclonal antibodies, and cytokines such as interferons and growth factors, as well as succinylcholine and other neuromuscular drugs that express quaternary ammonium epitopes that can cause such drugs to become multivalent.Citation3
Probably the clearest example of drug haptenation is that which occurs with penicillin which is chemically reactive and undergoes stable covalent binding to proteins or peptides, resulting in the creation of an immunogenic self-protein.Citation4 There is evidence that this often happens with albumin, perhaps because it is a common serum and tissue protein.Citation5,Citation6 The extent of haptenation that occurs may be dependent on the concentration of drug available but also depends on other factors, since fewer than 20% of available lysine molecules on albumin are modified by β-lactams. In the case of penicillins, we have considerable information on the penicillin derivatives that are responsible for sensitization and the production of the acute reaction.Citation7,Citation8 Penicillins bind to amino acids because the β-lactam ring opens spontaneously and forms stable covalently bound conjugates with proteins. This results in a penicilloyl epitope which is immunodominant in penicillin-specific immune responses. Haptenation can also occur through carboxyl and thiol groups to form minor determinants.
The related cephalosporins also can form hapten-protein conjugates, but this is slower and less efficient. It is presumed that a cephalosporyl determinant is generated between the β-lactam ring and lysine residues but the conjugate is unstable and fragments in the dihydrothiazine ring. These multiple breakdown products are not well defined, but it appears that IgE antibodies can bind to the side chains, to the β-lactam ring or the whole molecule.Citation9
Sulfamethoxazole is a drug which does not react covalently with proteins but becomes reactive through cytochrome P450 modification, first to sulfamethoxazole hydroxylamine and then by auto-oxidation to a nitroso intermediate which modifies thiol group on proteins.Citation2,Citation10 SMX-NO binds to non-MHC-associated protein, generating a protein conjugate that stimulates T-cell receptors via a hapten mechanism involving processing of the conjugate and liberation of antigenic peptide fragments (). N-acetyl transferase polymorphisms may play a role in determining this hypersensitivity response.Citation11 But sulfamethoxazole can also interact directly with antigen-presenting cell receptors.
Figure 1. MHC presentation of drug-haptenated peptide to the T cell receptor.
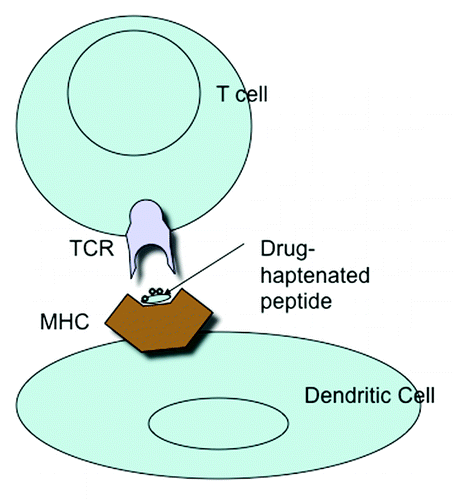
Processing Drug-Conjugates
The drug or a reactive product reacts with a self-protein or peptide which results in formation of a novel drug-conjugate or adduct. This undergoes antigen-processing to generate a small but novel MHC ligand that is loaded onto the MHC and transported to the cell surface, where it can interact with antigen-specific T cells (). This process requires a metabolically active antigen-presenting cell and time. Once generated, the MHC-drug-peptide complex is stable and ligand removal requires peptide exchange or peptide stripping from the MHC groove.Citation12
The P-I Concept
However, not all drugs seem to be capable of interacting covalently with proteins, and some immune reactions to drugs occur without antigen-processing. This has led to the pharmacological interaction or p-i hypothesis, whereby some chemically inert drugs are able to bind non-covalently to antigen-presenting structures such as the T cell receptor or MHC and cause stimulation directly of an immune response, as is the case with sulfamethoxazole.Citation13 Most drugs have been designed to fit into protein pockets in receptors and enzymes. This is the pharmacophore concept, based upon steric and electronic features that permit optimal interaction with a specific biologic target to trigger or block the target’s biologic response.Citation2,Citation8,Citation14 The p-i hypothesis has been derived from experimental work with T cell clones and TCR-transfected hybridomas expressing drug-specific receptors. The drug-interaction with the receptor is highly specific such that small changes in drug structure can affect reactivity. T cell activation can occur rapidly, before metabolism and processing of the drug could occur. However the reactions that occur are the same as those induced by a drug-modified peptide antigen, since the immune response once activated proceeds in a fixed way. The hypothesis could explain why reactions can sometimes occur without known previous sensitization
Evidence in favor of the p-i hypothesis is (1) aldehyde fixed antigen presenting cells (APC) are able to activate drug-specific T cell clones if incubated with the drug, (2) drug binding by this mechanism is labile and reversible, (3) calcium influx into T cell clones exposed to the drug occurs within seconds, (4) elution of peptides from HLA-B*1502, which presents carbamazepine to drug-reactive T cell clones, does not show the presence of covalently bound carbamazepine modified peptides.Citation15
In the case of sulfamethoxazole, interaction with the TCR occurs in the CDR2 and CDR3 regions of TCR Va and Vb, where there are presumably drug binding sites. But in addition, it has been proposed that drugs might bind to certain HLA-B alleles and the modified allele is then recognized by the TCR. Finally binding may first be to the TCR giving a partial activation followed by the TCR interacting with drug bound to MHC class II molecules, resulting in complete activation of the T cells ().Citation2,Citation16
Figure 2. Activation of T cell &/or Dendritic cell by non-covalent binding of drug to T cell receptor or MHC.
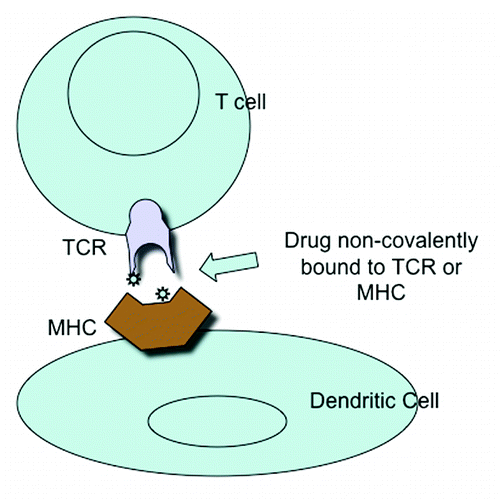
T cells from sulfamethoxazole-allergic patients can be stimulated with the parent drug sulfamethoxazole and SMX-NO bound directly to MHC molecules and speci□c T-cell receptors. Both the parent drug and drug metabolite apparently contain the steric and electronic features required to ensure molecular interactions with both immunologic receptors and stimulation of a T-cell response. It seems likely that only multivalent MHC interactions can induce structural rearrangement of the TCR so that Tyr rich regions are exposed.Citation2,Citation6 CD4/CD8 binding is not necessary for TCR activation but high affinity binding to the receptor is necessary. So drug binding may convert a low afinity antigen to a high affinity antigen.
While drugs have the potential to provide maturation signals to dendritic cells, acting as co-stimulatory agents, at the time when drugs are being administered, these cells may also receive maturation signals from other sources such as infections or other diseases or trauma. Lavergne et al.Citation17 demonstrated metabolism of sulfamethoxazole by dendritic cells, with adduct or conjugate formation being enhanced by bacterial endotoxins, viral proteins and cytokines. High levels of protein antigen were detected after 16 hours and antigen-presenting cells pulsed with sulfamethoxazole stimulated drug-reactive cells cloned from allergic individuals. This response was blocked by inhibition of metabolism in the antigen-presenting cells.
Therefore drugs as haptens can give two signals to dendritic cells. In lower concentrations there is partial activation to a semi mature dendritic cell phenotype. At higher concentrations there may occur bystander cell death and signals for full maturation of dendritic cells.
Amoxicillin has been shown to induce a semi mature state in dendritic cells, in which they can stimulate a T cell response. Sulphamethoxazole and its metabolites as well as abacavir have been shown to induce CD40 expression on dendritic cells.Citation18-Citation20 Drug-related maturation signals might also trigger reactivation of latent virus-specific CD8 cells in conditions such as DHIS.Citation21
Role of MHC
The role of the MHC in drug allergy has received particular attention with regard to certain drugs and ethnic groups. The drug hypersensitivity syndrome caused by abacavir is strongly associated with HLA-B*5701 and key structures in the peptide binding cleft of HLA-B*5701 have been identified that permit non-covalent interactions with this drug.Citation21-Citation23 This binding may also occur in so-called empty, non-peptide bearing MHC class I molecules. The binding of abacavir to the F pocket of HLA-B*5701 alters the spectrum of self peptides that can be presented by this MHC class I molecule. This results in the display of a novel peptide repertoire that appears foreign to the immune system. Memory T cell responses in abacavir-hypersensitive donors are directed against a self-peptide that requires abacavir to efficiently bind to HLA-B*5701. The situation is then analogous to alloreactions with the development of a severe cytotoxic response through the activation of cross-reactive effector memory cytotoxic T cells. The rapidity of onset of the reaction and its intensity may depend upon differences in TCR avidity.Citation24
However, abacavir also appears to bind irreversibly to proteins in antigen-presenting cells, raising the possibility that in this case, both non-covalent and altered peptides mechanisms may be involved in the generation of the allergic drug reaction.Citation25,Citation26 The association with HLA-B*5701 is strong enough that the US Food and Drug Administration issued an alert in 2008 recommending pre-screening for this allele before prescribing the drug.
In Han Chinese and other Asian populations, Stevens-Johnson/Toxic Epidermal Necrolysis induced by carbamazepine is strongly associated with HLA-B*1502.Citation25,Citation27,Citation28 This association does not exist in Europeans, where an association with HLA-A*3101 has been reported.Citation29 It does not appear that carbamazepine covalently modifies peptides which are presented in MHC but rather that carbamazepine or it metabolites binds directly to endogenous peptides already present there.Citation28
However, not all HLA-B*1502 bearing patients react in this way to carbamazepine. This appears to be due to the requirement for a particular structure of the T cell receptor.Citation30 T cell clones from HLA-B*1502 donors with carbamazepine hypersensitivity express the predominant TCR clonotype VB-11-ISGSY. This was found in 84% of 19 patients with SJS/TEN but was absent in carbamazepine-tolerant patients. It is present in 14% of healthy subjects. If peripheral blood mononuclear cells from normal donors expressing HLA-B*1502 and VB-11-ISGSY were primed with carbamazepine, a cytotoxic T cell response was induced that could be blocked with anti-TCR-VB-11. Thus the appropriate HLA allele and T cell receptor clonotrype is required for the carbamazepine immune response to occur in this population.
The severe mucocutaneous lesions of SJS/TEN are usually explained on the basis of cytotoxic T lymphocytes (CTLs)/ natural killer (NK) cell-mediated reactions.Citation31 Three major types of reactions have been considered responsible for these reactions, (1) Fas-FasL (Fas Ligand) interactions, (2) the perforin/granzyme B pathway, (3) granulysin. Viard et al.Citation32 proposed that Fas-FasL interactions were responsible for apoptosis of keratinocytes in SJS/TEN. However this hypothesis has been questioned and a role for perforin/granzyme B suggested.Citation33 Activated CTLs and NK cells produce perforin and this can open a channel in the target cell membrane to allow granzyme B to enter, which activates the caspase cascade. The 15-kDa granulysin is secreted by CTLs and NK cells and is present in blister fluid in SJS/TEN. This has a cytotoxic effect on keratinocytes and also acts as a chemoattractant for T cells, monocytes and other inflammatory cells.
Role of Viruses
Drugs are also frequently associated with viral infections and in most cases; the drug is blamed for the exanthema that occurs. Sometimes this is true and the patient has a persistent delayed-type allergy to aminopenicillins. But more often, the reaction does not recur on re-administration of the drug. The rash in this case may be caused by a lowering of the T cell threshold for drug reaction during the infection, or from infection-induced alterations in drug metabolism or virally-induced polyclonal T cell activation. Although Epstein-Barr virus infection is the best known example, other viruses similar predispose to drug-induced exanthema.
Drug-induced hypersensitivity syndrome (DIHS) is a multi-organ disorder characterized by cutaneous disease, eosinophilia and multi-organ failure occurring within 8 weeks after the introduction of a drug. The original description involved aromatic anticonvulsants and was termed “anticonvulsant hypersensitivity syndrome.” Later the name “drug rash with eosinophilia and systemic symptoms” (DRESS) was coined but this has been replaced by DIHS.
An intimate relationship appears to exist between viruses and the occurrence of DIHS.Citation34 In this syndrome, it was demonstrated by PCR analysis that there occurs a multiple and sequential reactivation of Herpes viruses in DIHS. Investigations have shown, by lymphocyte transformation tests and patch tests, that drug-specific T cells are involved in initiating the syndrome.Citation34 The drugs implicated in DIHS appear to be either chemically reactive, binding to antigen binding structures such as MHC or tissue proteins, or are metabolized to oxidative by-products that are reactive, or if chemically unreactive, like lamotrigine, bind non-covalently to the T cell receptor or MHC, as described in the p-I concept.Citation28 The occurrence of T cell activation may reactivate viral genome in the T cells leading to an immune response against the virus. Takahashi et al.Citation35 found an expansion of active T regulatory (Treg) cells during the acute phase of DIHS that might cause a sequential reactivation of Herpes viruses. As the condition begins to resolve, these cells lost their function, resulting in the potential for an increase in the risk of developing autoimmune disease.
Alternatively, the symptoms of DIHS may be due to both oligoclonal expansion of drug-specific T cells and virus-specific T cells that express cross-reactivity with drugs. Drug administration may expand these cross-reacting T cells, triggering latent virus reactivation. This might explain the association of relapses in the absence of the drug with virus reactivation, the involvement of multiple organs and the cross-reactivity with multiple drugs seen in DIHS. Of note, evidence of the viral genome is delayed 2–3 weeks from the start of the syndrome.Citation36 One possible explanation for this is that several of the causative drugs have the potential to cause immune suppression. So viral clearance may only occur after drug withdrawal and symptoms may reflect the return of the immune response to the virus.
Drug-Induced Liver Injury
The liver is the site of metabolism for many drugs and reactive metabolites are formed frequently, yet reactions involving the immune system do not occur often. Perhaps it is necessary that drug metabolites be capable of generating danger signals such as high mobility group box (HMGB) 1, heat shock proteins and S100 proteins from damaged cells, to act as costimulators. It is also possible that damaged hepatocytes release damage-associated molecular pattern (DAMP) molecules which stimulate a proinflammatory response in innate immune cells such as neutrophils and macrophages.Citation37 Such cells contain cytochrome 450 and/or peroxidase enzymes capable of generating reactive metabolites from drugs. Apoptotic and necrotic cells can also release DAMP molecules such as HMGB1 protein, heat shock proteins, hyaluran, surfactant protein, B-defensin and cardiolipin to act as stimulators of immune responses.
Halothane induced liver damage is characterized by initial mild liver injury, followed by, in a small percentage of patients, massive hepatocyte necrosis on re-exposure. An understanding of the mechanisms of this response is of importance, because of the reports of hepatitis with newer volatile anesthetics.Citation1,Citation38
A mouse model of halothane -induced liver injury has been described, particularly inducible in Balb/c mice.Citation37,Citation39 The model appears to be dependent upon formation of Trifluoroacetic acid-protein adducts but susceptibility is attributed to a differential activation of the innate immune system. After halothane exposure, there is an increase in proinflammatory cytokines such as TNF-a, IL-1B, IL-6 and iNOS. Evidence of an innate immune response was seen with increase of IL-8 and increased neutrophil infiltration in to the liver. A role for neutrophils in the induction of liver damage was confirmed by neutrophil depletion studies. Using wild type and NKT cell-deficient CD1−/− mice, it was shown that these cells were involved in neutrophil recruitment into the liver. Other studies have suggested a role for NK cells and cytokines IL10 and IL-17. Therefore the innate inflammatory response may be an important determinant of drug-induced liver disease.
In some cases drug-induced liver disease involves the adaptive immune system. Flucloxacillin causes cholestatic liver damage.Citation40-Citation42 The drug binds to lysine resides 190, 212 on human serum albumin and most T cell clones from flucoxacillin allergic patients are CD8+ Granzyme +. Flucloxacillin stimulation of these T cells is HLA class I dependent and may be HLA-B*5701 restricted. Studies showed a 3-fold increased risk of flucloxacillin hepatotoxicity in individuals with polymorphisms in the promoter region of the pregnane X receptor (PXR; also known as NR1I2), which is activated by flucloxacillin.
Classification of Drug Reactions
Allergic drug reactions are considered to be the result of drug recognition by the immune system with clinical manifestations that are not explainable by the known side effects of the drug. However frequently the pathophysiology of the reaction is unknown and it is considered to be allergic in nature because it happens infrequently or to certain populations and the presentation is consistent with an immunological response.
Attempts are usually made to fit the manifestations of allergic drug reactions into the classification of Gell and Coombs.Citation8,Citation43
In this classification, Type 1 reactions are immediate, or IgE mediated, Type 2 reactions are caused by IgG or IgM antibodies directed to cell surface antigens, Type 3 reactions involve IgG antibodies against soluble antigens, forming immune complexes, and Type 4 reactions are cell-mediated, subdivided as a, b, c and d.
Type 4a reactions are typified by the tuberculin skin reaction but may also participate in contact dermatitis and some bullous exanthema. They are considered to be Th1 reactions mediated by IFN-γ and TNF-α and monocyte and macrophages are the primary effectors. Type 4b reactions are exemplified as Th2 reactions, with IL-5, IL-4 and IL-13 and eotaxin driving eosinophilic inflammation. Examples include maculopapular and bullous exanthema. In Type 4c reactions, cytotoxic T cells are the primary mediators with perforin, granzyme B and FasL release causing contact dermatitis, maculopapular and bullous eruptions. Type 4d reactions involve T cells and CXCL8 and GM-CSF causing neutrophilic infiltrates leading to pustular exanthema.
While there are individual examples of drugs that cause allergic reactions conforming to this classification, for many there is no definite proof that a particular mechanism is involved or multiple mechanisms can be implicated.
Immediate drug reactions or Gell and Coombs Type 1 reactions are the result of the cross-linking of drug-specific IgE molecules bound to mast cells and/or basophils. To do this, drugs must exist in a multivalent form expressing several epitopes and likely therefore bound to a peptide or protein molecule. If the drug is in its original state, expressing only a single epitope, cross-linking cannot occur. The exception would be drugs that can exist in a polymerized state in vivo such as succinylcholine and possibly aminopenicillins. When cross-linking occurs, mast cell degranulation results in the release of inflammatory mediators such as histamine; proteases (tryptase, chymase, carboxypeptidase); proteoglycans (heparin, chondroitin sulfate E);prostaglandins (PGD2); leukotrienes (LTC4); cytokines (TNF-α, IL-3, IL-4, IL-5, IL-6, IL-8, IL-16, GM-CSF);chemokines (CCL2, CCL3, CCL11).Citation44 These factors cause the typical signs of anaphylaxis such as urticaria and angioedema as well as bronchospasm, hypotension and cardiac arrhythmias. While all drugs can potentially cause immediate reactions, β-lactam antibiotics are a common cause, as well as muscle relaxants.
For other low molecular weight drugs causing immediate reactions we know almost nothing about the epitopes involved unless they are demonstrable by skin or in vitro testing with the intact molecule.
Type 2 reactions are typified most commonly by hemolytic anemia, thrombocytopenia and neutropenia, in which drug or active drug metabolites bind to cell membranes and these epitopes become the target of drug-specific IgG or IgM antibodies, with subsequent complement activation and either hemolysis or phagocytosis in the reticulo-endothelial system. Again, penicillins are recognized to cause such reactions but so can cephalosporins, β-lactamase inhibitors and methyl-dopa.Citation45
Type 3 reactions are considered to be mediated by immune complexes, which requires the drug or its active metabolites to be covalently bound to a macromolecule such as albumin. An immune response to the drug-conjugate, usually in the form of IgG antibodies, then results in the formation of immune complexes that deposit in such organs as skin, kidney and joints. Since complement is activated, there is release of anaphylatoxins such as C3a and C5a, with induction of mast cell mediator release and attraction of neutrophils to the site of immune complex deposition. There may also be stimulation of an IgE response with subsequent mast cell activation. Such reactions are delayed for 8–10 days during the primary sensitization, but if the drug is readministered in a sensitized individual, the reaction is accelerated, occurring within 2 d of restarting the drug. Penicillins are again often associated with these reactions, as are cephalosporins and macrolides but commonest now as a cause are drug macromolecules such as monoclonal antibodies.Citation43,Citation46
Many drugs are capable of inducing Type 4 reactions, which are believed to be the cause of most maculopapular eruptions. Some of these may be Type 4b and others Type 4c, a classification dependent on the histology of the rash or in vitro studies of the types of T cells involved and cytokines released. Most contact reactions to drugs appear to be either Type 4a or Type 4c.
Acute generalized exanthematous pustulosis is characterized by the development of acute, small, non-follicular sterile pustules beginning in the intertriginous folds with diffuse edema and erythema. There is fever and a peripheral leukocytosis. Histologically there occurs massive subcorneal infiltration with neutrophils and T cell infiltration of the dermis and epidermis. When drug-induced, the lymphocyte transformation test shows marked reactivity to the causative drug. Drug-specific CD4 and CD8 positive T cells are found in the peripheral blood and these produce interleukin-8 (IL-8/CXCL8). The neutrophil infiltration may therefore be secondary to these cells infiltrating the skin, or drug-specific T cells may stimulate keratinocytes to produce IL-8. Another possibility is that IL-17-producing Th17 cells that co-express IL-22 are responsible for synergistically stimulating IL-8 production by keratinocytes.Citation16,Citation47
Allergic responses to drugs that involve the adaptive immune system are dependent upon the activation of three different types of effector T helper cells, these being Th1, Th2 and Th17.Citation48,Citation49 While Th1 cells produce predominantly IFN-γ and generate a cell-mediated immune response, Th2 cells produce IL-4, IL-5 and IL-13 and stimulate IgE production, resulting in immediate Type 1 reactions, and Th17 cells are responsible for neutrophilic responses mediated through IL-17.
Differentiation of these Th cells is directed by antigen-presenting cells, most often dendritic cells. Dendritic cells can be activated by tissue damage and cell death, as well as by some drugs, which explains why allergic drug reactions frequently occur in association with viral, bacterial or other infections.Citation17,Citation50In the case of drug hapten-peptide conjugates, it remains uncertain what characteristics determine the type of Th cell activated. It is clear from clinical experience that penicillin is capable of inducing IgE, IgG and IgM responses, as well as cell-mediated responses, causing Type 1, Type 2, Type 3 or Type 4 reactions. What factors determine which of these immune responses develops? In the case of Type 1 reactions, there is interesting data showing that basophils and mast cells can act as APC and induce Th2 responses.Citation51 In circumstances where Type 2 reactions occur there has often been prolonged treatment with high dose β-lactam antibiotics. And in Type 3 reactions, there may be unique properties of the IgG antibodies specific for the drug hapten-protein conjugate that results in immune complex deposition in tissues, or there may occur concomitant generation of drug-specific IgE antibodies. For Type 4 reactions, in situations where T cell activation is via pharmacological interaction, responses will be T cell mediated, although this does not preclude Th cell activation and antibody production. In Type 4d reactions, Th17 cells may be involved, while Type 4c reactions may involve Th2 cells. Type 4b reactions involve cytotoxic T cells initially activated by Th1 helper cells.
Interesting studies by Pichler and his collaborators in Multiple Drug Hypersensitivity suggest a problem exists with persistent T cell activation in such patients, even in the absence of drug, perhaps implying in vivo activation by chronic viral infections such as Herpes virus, CMV, EBV etc.Citation52 In such patients there does not seem to be a deficiency in Treg activity.
Drugs are often recognized as foreign by the immune system even in the absence of clinical hypersensitivity, so IgG and IgM antibodies to drugs may be found. So, what turns an innocuous immune response to a drug into clinical hypersensitivity? While a number of molecular characteristics of allergens have been described, such a < 70 kd size, low hydrophobicity, high stability, lack of bacterial protein sequences and the presence of allergen-specific patches with surface-exposed hydrophobic residues, none clearly define allergenicity and most would not apply to drug-modified self-proteins, although they could apply to drug-modified proteins from infectious agents.Citation53,Citation54 Development of drug allergy may indeed be because of other factors activating the innate immune system, such as viruses or bacteria, or the unique characteristics of the individual, including the presence of certain HLA alleles or T cell receptor structures that allow drug hypersensitivity to develop. However, the fact that individuals previously allergic to a drug can often tolerate the drug subsequently indicates that environmental influences are important.
Some drug reactions appear to conform to a Type 1 pattern with urticaria, angioedema and often bronchospasm, but actually do not involve the adaptive immune system at all. Examples of these would be reactions to narcotics, which stimulate direct histamine release from mast cells or basophils, certain antibiotics such as vancomycin, producing the “red man” syndrome by direct histamine release, and reactions to NSAIDS. The cause of the latter is still uncertain, with polymorphisms in cytochrome P4502C9, cyclo-oxygenase and lipoxygenase and thromboxane A2 receptor being associated with ASA-induced urticaria.Citation55,Citation56 Aspirin exacerbated respiratory disease (AERD) is a reaction similar to a Type I reaction but without production of IgE by the immune system, related to excessive production of leukotrienes by 5-lipoxygenase, which results in mast cell degranulation and release of mast cell mediators and cytokines, causing symptoms of rhinitis and asthma.Citation57,Citation58 Prostaglandin E2, produced via the cyclo-oxygenase pathway, is an inhibitor of 5-lipoxygenase. Aspirin inhibits production of PGE2, allowing increased leukotriene production. Reduced PGE2 synthesis also increases mast cell instability with enhanced release of histamine and other mediators. There is also evidence that IL-10 and TGF-b polymorphisms play a role in AERD. Angioedema is precipitated by Angiotensin converting enzyme inhibitors and although relatively rare, can be severe.Citation59 Angiotensin converting enzyme catalyzes the conversion of Angiotensin I to Angiotensin II, but also degrades bradykinin, when the same enzyme is known as kininase II or dipeptidylcaboxypeptidase I. Inhibition of kininase II prevents degradation of bradykinin and other kinins, predisposing to development of angioedema. An association exists between low plasma levels of aminopeptidase P and angioedema because this enzyme is important in bradykinin metabolism when Angiotensin-converting Enzyme (ACE) is inhibited.
Common Causes of Drug Reactions
Penicillins
Penicillins are the most frequent cause of drug allergy, reportedly affecting about 10% of patients. However 90% of patients claiming to be allergic to penicillin will have negative skin tests for penicillin allergy. The commonest reactions associated with penicillins are anaphylaxis, urticaria and angioedema, or maculopapular rashes. Carbapenems do not exhibit a significant degree of cross reactivity with penicillin and may be administered in penicillin-allergic subjects after prophylactic skin tests with the relevant Carbapenem. Monobactams such as aztreonam are well tolerated in patients with penicillin allergy, but patients with an allergy to ceftazidime should avoid aztreonam. Second or third generation cephalosporins have a very low degree of cross-reactivity in penicillin allergic subjects. If penicillins are essential in a patient with proven penicillin allergy, then desensitization should be performed.Citation60,Citation61
Cephalosporins
Cephalosporins most commonly cause maculopapular rashes and drug fever, with urticaria less commonly seen and anaphylaxis rare. Positive skin tests to penicillin are associated with a small increased risk of reacting to first generation cephalosporins. In those individuals with cephalosporin allergy, there is a very limited cross-reactivity between second and third generation cephalosporins and penicillins, particularly amino-penicillins, based on side chain similarities. In such patients, skin testing with the proposed antibiotic to be used is useful, or a graded challenge. In the presence of positive skin tests with the proposed drug, if there is no alternative, desensitization should be performed.Citation9
Sulfonamides
Sulfonamide antibiotics are often responsible for delayed maculo-papular rashes and cause SJS and TEN. Patients with HIV are more likely to develop rashes from sulfonamide antibiotics, perhaps because of chronic immune stimulation and frequent exposure. Since Trimethoprim-sulfamethoxazole is often required to treat HIV-associated infections, induction of tolerance to TMP-SMX is often required. It should be noted that the chemical structure of non-antibiotic sulfonamides such as thiazide diuretics, some NSAIDS and anticonvulsants lacks the required antigenic determinants to induced an immune response in those with TMP-SMX allergy and usually can be safely administered, except for sulfasalazine, which is degraded in the intestine to sulfapyridine, with an aromatic immunogenic determinant like sulfamethoxazole.Citation62,Citation63
Acetylsalicylic acid/NSAID reactions
Acetylsalicylic acid (ASA) and NSAIDS may be responsible for allergic reactions involving the adaptive immune system, but more frequently cause pseudo allergic reactions resulting from abnormalities of prostaglandin or leukotriene synthesis. Reactions include exacerbation of respiratory disease such as asthma or symptoms of nasal polyposis, urticaria, angioedema, and anaphylaxis. Almost all of these reactions are caused by cyclo-oxygenase 1 (cox-1) inhibitors. Such patients should carefully avoid cox-1 inhibitors unless they have been desensitized. Selective cox-2 inhibitors almost never cause such reactions and can be safely taken by patients with ASA/NSAID allergy.Citation64
When urticaria/angioedema are caused by cox-1 inhibitors, it may be precipitated by all or some of that group of drugs. For example, ASA-sensitive patients may react to ibuprofen and naproxen but not to other cox-1 inhibitors. Patients with chronic urticaria often have exacerbations if they take a cox-1 inhibitor. However true allergic reactions to NSAIDS involving the adaptive immune response, such as those occurring to diclofenac are usually drug-specific and other cox-1 inhibitors are tolerated.Citation65
Angioedema from ACE-inhibitors
Although angioedema from ACE-inhibitors is uncommon, developing in 0.1–2.2% of patients on ACE-inhibitors, it accounts for about one third of emergency room cases of angioedema. Such patients often end up being admitted to hospital and requiring intensive care management.Citation59 Angioedema from ACE-inhibitor may occur soon after beginning therapy but can also occur after months or years of treatment. Risk factors, in addition to hereditary angioedema, include older age, female sex, smoking, NSAID use and ACE-inhibitor-induced cough. Concomitant treatment with statins and sirolimus is a risk factor. Because the angioedema is induced by kinins, it is not responsive to usual treatments such as epinephrine, anti-histamines and steroids. The new bradykinin receptor 2 antagonist Icatibant is likely to be effective.Citation66
General anesthetics
Anaphylaxis occurs in patients undergoing general anesthesia. Investigation of such reaction is difficult because the patient is often exposed to multiple drugs and other agents administered over a short period of time. Reactions in such circumstances are often due to neuromuscular blocking agents, but have been reported with intravenous anesthetics such as propofol, thiopentone and etomidate, as well as antibiotics, NSAIDS and latex. Inhaled anesthetics are not found to be responsible. Chlorhexidine and ethylene oxide have been recently incriminated in intra-operative anaphylaxis.Citation67
Allergic reactions to biological agents
Chimeric monoclonal antibodies such as infliximab, because they contain variable regions that are murine in origin, can stimulate an immune response. The presence of such antibodies varies from 12–60%, dependent upon the population studied. Humanized monoclonal antibodies such as alemtuzumab can induce immune responses in as many as 23% of patients with multiple sclerosis and fully human antibodies such as adalumumab are immunogenic in 3–4% of patients with Crohn’s disease and even higher levels are seen in Rheumatoid arthritis patients. Recombinant biologics such as abatacept and etenercept elicit antibody responses in up to 18% and 3% of patients respectively. The frequency of antibody production to biologics clearly varies with the populations studied. For example, rituximab induces antibodies in 1% of patients with B cell malignancies, 25% of patients with Sjogren’s syndrome and 40% of patients with SLE.Citation68
Most anti-drug antibodies are IgG in isotype but a proportion of reactions have been associated with IgE antibodies. These have been found to occur with infliximab, tocilizumab, cetuximab, natalizumab and muromonab, as well as rituximab and trastuzumab.
A number of infusion reactions occurring with monoclonals are the result of massive cytokine release by cells such as monocytes, macrophages, T cells, B cells and NK cells. This is an acute phase reaction indistinguishable from an IgE mediated reaction but occurring as a result of the first administration.
Stevens-Johnson and toxic epidermal necrolysis
The commonest drug causes of SJS and TEN are sulfonamides, allopurinol, carbamazepine, phenytoin and NSAIDS of the oxicam type. New drugs with increased risk are nevirapine and lamotrigine.Citation27,Citation31,Citation69
Although a strong association in Han Chinese has been shown for HLA-B*1501 and SJS, this has only been duplicated in a Thai population and in Europe, this allele is not a marker for SJS caused by carbamazepine, sulfonamides, lamotrigine or oxicam-NSAIDS. In Han Chinese HLA-B*1501 is associated with phenytoin and lamotrigin- induced severe cutaneous reactions.
A second association has been reported for allopurinol in the case of HLA-B*5801 in Han Chinese, Thai patients and Europeans.
Drug-induced hypersensitivity syndrome
This syndrome is characterized by an extensive mucocutaneous rash, fever, lymphadenopathy, hepatitis, eosinophilia and atypical lymphocytosis with damage to kidneys, heart, lungs and pancreas.Citation21 It occurs 3 weeks to 3 mo after beginning drug therapy and may persist or worsen after withdrawal of the medication. It is rare but more common than SJS. It is seen most commonly with anticonvulsants, antidepressants, sulfonamides and sulfones, NAIDS, anti-infective agents, Angiotensin converting enzyme inhibitors and betablockers. The most typical forms of DIHS occur with aromatic anticonvulsants, dapsone, sulfapyridine, allopurinol and minocycline. As noted previously, studies suggest a close relationship between DIHS and reactivation of Herpes 6 virus.
Drug-induced liver injury
Drug-induced Liver Injury (DILI) can be caused by drugs such as halothane, tienilic acid, hydralazine, diclofenac and carbamazepine where the reaction occurs within 1–8 weeks, is associated with rash, fever, eosinophilia and recurs rapidly on re-challenge.Citation70 There may also be present specific antibodies against hepatic proteins either native or drug-modified. There is also increasing evidence of an association between DILI and certain MHC class I and II alleles where the drugs involved are amoxicillin-clavulinate, flucloxacillin, ticlopidine, ximelagatran, lumiracoxib and lapatinib.
Desensitization in Immediate Allergy
Drug desensitization is indicated when (1) no alternative drug is available, (2) the drug is more effective than alternatives, (3) there are no co-morbid factors placing the patient at increased risk, (4) the previous reaction to the drug was not a severe, life-threatening immunotoxic reaction, vasculitis or bullous skin disease such a SJS/TEN or DIHS.Citation71,Citation72 Desensitization in general should be performed by a physician trained in management of anaphylaxis, the exception being when dealing with non-immediate reaction. An intravenous line and continuous monitoring is obligatory for immediate reactions. As a general rule, protocols should be used that have been applied to samples larger than 10 patients. Most protocols begin with a dose range from 1/10,000 to 1/100 of the full therapeutic dose. This is determined by the severity of the initial reaction. Doses are doubled every 15–20 min either by oral or intravenous administration until a therapeutic dose is reached. The oral route seems safer. Some protocols use 10-fold increases in dose but are associated with more side effects. Betablockers must be discontinued. For most protocols, except for chemotherapy regimens, pretreatment with antihistamines and steroids is not indicated. Validated methods of desensitization are limited to certain categories of drugs and certain types of reactions (). However anecdotal reports of desensitization for many types of drug allergy are reported, usually involving very limited numbers of patients. A website-based database of drug allergy case reports and desensitization protocols would therefore be of value.
Table 1. Common drug allergies and desensitizations
Animal models
Woo et al.Citation73 developed a murine model of systemic anaphylaxis to penicillin V. These allergic mice were desensitized by oral feeding with the drug, by five feedings of doubling doses every 15 min. Desensitization was determined by testing for the induction of active systemic and active cutaneous anaphylaxis and by measurement of plasma levels of platelet-activating factor and histamine. Mice fed more than 3 mg of a cumulative dose of penicillin V were completely protected from fatal systemic anaphylaxis but the desensitized state lasted only for about one hour. Antigen-specific mast cell desensitization appeared to be the mechanism for the desensitized state and not hapten inhibition, consumption of IgE or depletion of mast cell mediators. The authors suggested that the desensitization might result from the internalization of penicillin-specific IgE antibodies and that subsequent recycling of the internalized antibodies could lead to a recurrence of anaphylactic sensitivity.
Park et al.Citation74 investigated the effect of anti-IL-4 antibody in a murine model of penicillin V and cephalothin anaphylaxis. The IgE mediated state resulted in fatal reactions in 100% and 70–90% of animals with penicillin V and cephalothin readminstration. Administration of anti-IL-4 at the beginning of the sensitization procedure completely prevented anaphylaxis. But anti-IL-4 was also effective for 21 days in preventing fatal anaphylaxis when given to previously sensitized mice. This was associated with a fall in specific IgE levels in the treated animals. This has not been tested in humans.
Effects on mast cell and basophils
It is thought that mast cells and perhaps basophils are the targets for desensitization in immediate reactions to drugs in humans. By incremental administration of increasing amounts of drug, hypo-responsiveness of these cells is induced without anaphylaxis occurring. The tolerization in the individual patient is temporary.Citation75
The desensitization process may (1) deplete the cells of activating signal transduction components such as syk kinase, (2) depletion of mediators, (3) internalization of FceRI through progressive cross-linking at low antigen concentration.
There is evidence that the desensitized state persists as long as antigen is present in the environment but mediator release can occur again when antigen is removed. Signaling pathways thus remain intact for a second stimulus. In addition, FcεRI is not depleted from the mast cell surface. A fourth possibility that may apply in certain circumstances that would explain the ability to induced degranulation with a second allergen is haptenation of the specific IgE on the cell surface, such that cross-linking cannot occur.
In human basophils downregulation of FceR1 occurs during IgE activation but takes many hours to days. Three mechanisms may play a role in the termination of secretory signals in basophils. These are activation of SHIP, processing of syk by ubiquination and the degradation and loss of FceR1 receptors. A fourth mechanism, undefined as yet, involves resorting of receptors in the cell membrane. Two of these mechanisms appear to be the result of weak signaling. Downregulation of syk occurs with concentrations of allergen that do not induce secretion. Loss or downregulation of FceR1 can be demonstrated but does not occur with subthreshold stimulation.Citation75
The studies of MacGlashanCitation76 demonstrated that subthreshold desensitization of human basophils results in loss of syk and loss of cell surface receptor. These changes occur progressively over an extended period of time, without significant histamine release. It is assumed that during subthreshold stimulation, even though histamine release is not occurring, there is still present some signaling that mediates desensitization. Receptor loss appears not to be sensitive to the activities of syk, but is sensitive to the activities of src-family kinases. So loss of syk and loss of receptor expression may be disconnected under appropriate conditions.
Of considerable interest is the finding that when BPO2 and BPO(12)-HSA are compared in their ability to induce syk loss in basophils sensitized with BPO-specific IgE, there was only a small difference in the loss of syk for similar loading of the cell, even though histamine release was considerably different. It therefore seems that aggregate size is not a strong determinant of syk loss.
Benzylpenicilloylformyl-lysine or Ro-6-0787, a single epitope of the major antigenic determinant of benzylpenicillin, was used by De Weck and his collaborators to desensitize patients with penicillin allergy by haptenatin of IgE in a multicenter study of 90 patients.Citation77 Ro-6-0787 alone was successful in 17 of 26 patients, and combined with penicillin therapy, was successful in 42 of 46 cases. There was after its’ administration a depression of skin test reactivity to PPL and/or penicillin derivatives. But its use was unsuccessful in 11 cases and 5% of penicillin allergic subjects showed positive skin test reactions to this monovalent hapten. This study would appear to show that in some cases of penicillin allergy, that hapten inhibition of IgE is a potential cause of desensitization, but such a strategy would not be effective in those patients who were allergic to minor determinants.
Desensitization in NSAID sensitivity
Stevenson et al.Citation78reported in 1980 that two patients, who became refractory to ASA after oral challenge, noticed subsequent improvement in nasal obstruction and ability to smell. A randomized double blind placebo-controlled crossover trial of ASA desensitization in 25 patients with ASA-sensitive asthma subsequent showed significant improvement in symptoms of rhinosinusitis and decrease in daily nasal corticosteroid use while on ASA, but no effect on asthma was noted.Citation79 A retrospective review of 107 patients with ASA sensitivity with rhinosinusitis compared 65 patients who were desensitized to ASA compared with 42 patients who simply avoided all NSAIDS.Citation80 There were significant reductions in number of hospitalizations, ER visits, outpatient visits, sinusitis and sinus operations in the intervention group. There was also a reduced need for systemic steroids.Citation81 Subsequent longer term studies have shown benefit of ASA desensitization in terms of sinus symptoms, medication use and need for endoscopic sinus surgery, with operations reduced from every 3 years on average to every 9 years.Citation82
While the above studies describe the effects of oral desensitization with ASA, other methods have included local desensitization using l-lysine aspirin. This has been carried both intranasally and by bronchial inhalation.Citation83,Citation84
In patients with ASA-induced asthma and rhinosinusitis, after ASA-lysine desensitization, the number of inflammatory cells in the nasal tissues expressing Cys-LT1 receptors became normal after 5 months of therapy. Production of thromboxane B2 by monocytes was reduced and LTB4 synthesis normalized after chronic ASA therapy following desensitization. LTE4 levels in urine were reduced, but not to normal levels.
ASA has been shown to inhibit activation of transcription factor-kB to inhibit IL-4 and IL-13 activation of STAT-6.Citation85 So after desensitization, ASA may inhibit the inflammatory response as well as affecting prostaglandin and leukotriene production. But the precise mechanism by which ASA desensitization produces benefit remains unknown
Desensitization in Cell-mediated Reactions
Animal models
Phenytoin is a widely used anticonvulsant for treatment of focal and generalized tonic clonic seizures. It induces maculopapular rashes as well as SJS, TEN, exfoliative dermatitis and fixed drug eruptions. It is also responsible for DIHS with fever, rash and lymphadenopathy. In such delayed reactions, identification of allergen-specific T cells may be important. This may be achieved by patch testing or delayed skin testing, or by lymphocyte transformation testing, but these methods lack sensitivity and false negative tests are common. Recently, Turcanu et al.Citation86 described a carboxyfluorescein succinimidyl ester (CFSE) for identification of food allergen-specific T cells. This approach has been applied to patients with reactions to phenytoin to examine frequency and cytokine-producing phenotypes of CD4+ and CD4- T cells.
Phenytoin-specific proliferation and cytokine production was shown by the CFSE dilution assay with a sensitivity of 100% for proliferation in the CFSE assay and 71.4% for IFN-γ production.Citation87 Both CD4+ and CD4- cells responded with production of IFN-γ and TNF-α.
In a patient with phenytoin allergy, manifest by maculopapular rash appearing after 12 days therapy, which was treated with clinical desensitization, an effect of desensitization was shown on phenytoin responsive CD4+ and CD4- T cells.Citation88 Before desensitization, these numbered 3.09% and 3.18% respectively, by CFSE assay, with IFN-γ-positive cells 13.6% and 12.3% respectively. But after desensitization, no IFN-γ producing cells were demonstrable and phenytoin responsive cells in the CFSE assay were 0.16% and 1.26% in the CD4+ and CD4- T cell populations. The mechanism for this desensitization process and how these drug-specific T cells are tolerized or inhibited is unclear.
A possible explanation might be through the activation of T regulatory cells.Citation89,Citation90 Although there is no direct evidence for this, it is of interest that fixed drug reactions, where drug-specific CD4+ and CD8+ T cells have been demonstrated, the amelioration of the skin lesions coincides with an influx into the skin of Foxp3+ regulatory T cells. Teraki and ShioharaCitation91 showed in a patient with allopurinol fixed drug reaction that desensitization was associated with an influx of CD4+ CD25+ T cells into the skin lesions. Because the severe form of fixed drug reactions mimics TEN, these findings may be of clinical importance in determining new therapies for TEN of SJS and may be of relevance to our understanding of the mechanisms underlying desensitization in delayed drug reactions.
Desensitization in presumed delayed cell-mediated reactions in patients has been performed in individuals who have developed morbilliform reactions to drugs such as sulfamethoxazole and allopurinol as well as in fixed drug reactions.Citation92,Citation93 These protocols follow the general principles of rapid desensitization but are performed over a longer period, varying from two days to eight days. In the case of sulfamethoxazole, the desensitization is successful more than 80% of the time, depending upon patient selection. Demoly et al.Citation94 have recently described a six hour graded challenge that was successful in 95% of patients. However the mechanism involved in the desensitization, which presumably involves induction of some form of T cell tolerance or anergy, remains unknown.
Desensitization Allergy to Chemotherapeutic Agents
Castels and associates have developed protocols for desensitization with chemotherapeutic agents in patients who have suffered a Type 1 immediate hypersensitivity reaction, whether IgE or non-IgE mediated.Citation46,Citation75 The protocol is 12 to 20 step based, with doubling doses starting at 1/1000 of the final dose. This protocol, which has been used for paclitaxel and docetaxel, cisplatin, carboplatin, oxaliplatin and monoclonals such as rituximab, infliximab, trastuzumab, omalizumab, natalizumab, basiliximab, abciximab and cetuximab, is preceded by routine premedication with diphenhydramine and famotidine, which may be supplemented with aspirin, montelukast or glucocorticoids. Beta-adrenergic antagonists are discontinued for 24 hours. Desensitization with taxanes is generally well tolerated, with a series of 98 patients undergoing 413 desensitizations with only mild or no reactions. The success of rapid desensitization with platins appears to be similar. For monoclonal antibody desensitization, reaction rates are approximately 30% with 90% of these being mild. In the small percentage of cases with severe reactions, these occurred in patients with positive skin tests to the agent that was administered.
Summary
While we have learned a great deal about the pathogenesis of allergic drug reactions in terms of immediate allergy to penicillins and the role of MHC and viruses in some serious forms of drug allergy, such as SJS/TEN and DIHS, we know much less about the immunological processes involved in common drug reactions such as maculo- papular eruptions. Therefore our diagnostic techniques for demonstrating the presence of delayed drug allergy are severely limited and we lack an understanding of the mechanisms of desensitization in such situations. In the future, animal models may help to clarify these issues but, as is seen in SJS/TEN and DIHS, there are very specific requirements that must be met for drug allergy to occur which it may not be possible to model. More probable is that the development of sophisticated immunological techniques will allow careful dissection of the drug-immune response in human. Of great importance is the ability to develop predictive models that will allow the identification of patients at high risk of adverse reactions to drugs. We should not be surprised by the complexity of this problem, given system did not evolve to deal with multiple man-made chemicals, that the human immune.
| Abbreviations: | ||
| ACE | = | angiotensin-converting enzyme |
| AERD | = | aspirin exacerbated respiratory disease |
| APC | = | antigen presenting cell |
| ASA | = | acetyl salicylic acid |
| CFSE | = | carboxyfluorescein diacetate succinimidyl ester |
| CTL | = | cytotoxic T lymphocytes |
| DAMP | = | damage-associated molecular pattern |
| DIHS | = | drug-induced hypersensitivity syndrome |
| DILI | = | drug-induced liver injury |
| DRESS | = | drug rash with eosinophilia and systemic symptoms |
| FasL | = | fas ligand |
| FcεRI | = | high affinity IgE receptor I |
| HLA | = | human leukocyte antigen |
| MHC | = | major histocompatibility complex |
| NK | = | natural killer |
| NKT | = | natural killer T cell |
| NSAID | = | non-steroidal anti-inflammatory drug |
| PCR | = | polymerase chain reaction |
| PXR | = | pregnane X receptor |
| SHIP | = | SH2-containing inositol phosphatase |
| SJS | = | Stevens-Johnson syndrome |
| TCR | = | T cell receptor |
| TEN | = | toxic epidermal necrolysis |
| Th | = | T helper |
Related Research Data
References
- Pirmohamed M, Breckenridge AM, Kitteringham NR, Park BK. Adverse drug reactions. BMJ 1998; 316:1295 - 8; http://dx.doi.org/10.1136/bmj.316.7140.1295; PMID: 9554902
- Pichler WJ, Naisbitt DJ, Park BK. Immune pathomechanism of drug hypersensitivity reactions. J Allergy Clin Immunol 2011; 127:Suppl S74 - 81; http://dx.doi.org/10.1016/j.jaci.2010.11.048; PMID: 21354503
- McKinnon RP, Wildsmith JA. Histaminoid reactions in anaesthesia. Br J Anaesth 1995; 74:217 - 28; http://dx.doi.org/10.1093/bja/74.2.217; PMID: 7535078
- Idsoe O, Guthe T, Willcox RR, de Weck AL. Nature and extent of penicillin side-reactions, with particular reference to fatalities from anaphylactic shock. Bull World Health Organ 1968; 38:159 - 88; PMID: 5302296
- Meng X, Jenkins RE, Berry NG, Maggs JL, Farrell J, Lane CS, et al. Direct evidence for the formation of diastereoisomeric benzylpenicilloyl haptens from benzylpenicillin and benzylpenicillenic acid in patients. J Pharmacol Exp Ther 2011; 338:841 - 9; http://dx.doi.org/10.1124/jpet.111.183871; PMID: 21680886
- El-Ghaiesh S, Monshi MM, Whitaker P, Jenkins R, Meng X, Farrell J, et al. Characterization of the antigen specificity of T-cell clones from piperacillin-hypersensitive patients with cystic fibrosis. J Pharmacol Exp Ther 2012; 341:597 - 610; http://dx.doi.org/10.1124/jpet.111.190900; PMID: 22371438
- Levine BB. Immunologic mechanisms of penicillin allergy. A haptenic model system for the study of allergic diseases of man. N Engl J Med 1966; 275:1115 - 25; http://dx.doi.org/10.1056/NEJM196611172752009; PMID: 5923027
- Pichler WJ. Immune mechanism of drug hypersensitivity. Immunol Allergy Clin North Am 2004; 24:373 - 97, v-vi; http://dx.doi.org/10.1016/j.iac.2004.03.012; PMID: 15242717
- Perez-Inestrosa E, Suau R, Montañez MI, Rodriguez R, Mayorga C, Torres MJ, et al. Cephalosporin chemical reactivity and its immunological implications. Curr Opin Allergy Clin Immunol 2005; 5:323 - 30; http://dx.doi.org/10.1097/01.all.0000173788.73401.69; PMID: 15985814
- Elsheikh A, Castrejon L, Lavergne SN, Whitaker P, Monshi M, Callan H, et al. Enhanced antigenicity leads to altered immunogenicity in sulfamethoxazole-hypersensitive patients with cystic fibrosis. J Allergy Clin Immunol 2011; 127:1543 - 51, e3; http://dx.doi.org/10.1016/j.jaci.2010.12.1119; PMID: 21354601
- Wang D, Para MF, Koletar SL, Sadee W. Human N-acetyltransferase 1 *10 and *11 alleles increase protein expression through distinct mechanisms and associate with sulfamethoxazole-induced hypersensitivity. Pharmacogenet Genomics 2011; 21:652 - 64; http://dx.doi.org/10.1097/FPC.0b013e3283498ee9; PMID: 21878835
- Zaheer-ul-Haq, Khan W. Molecular and structural determinants of adamantyl susceptibility to HLA-DRs allelic variants: an in silico approach to understand the mechanism of MLEs. J Comput Aided Mol Des 2011; 25:81 - 101; http://dx.doi.org/10.1007/s10822-010-9404-y; PMID: 21116684
- Pichler WJ. Pharmacological interaction of drugs with antigen-specific immune receptors: the p-i concept. Curr Opin Allergy Clin Immunol 2002; 2:301 - 5; http://dx.doi.org/10.1097/00130832-200208000-00003; PMID: 12130944
- IUPAC recommendation on nomenclature and symbols. Clinical Chemistry Division. Commision on Toxicology. Glossary of terms used in toxicology synopsis. Acta Pol Pharm 1992; 49:1; PMID: 16092424
- Uetrecht J. Idiosyncratic drug reactions: current understanding. Annu Rev Pharmacol Toxicol 2007; 47:513 - 39; http://dx.doi.org/10.1146/annurev.pharmtox.47.120505.105150; PMID: 16879083
- Posadas SJ, Pichler WJ. Delayed drug hypersensitivity reactions - new concepts. Clin Exp Allergy 2007; 37:989 - 99; http://dx.doi.org/10.1111/j.1365-2222.2007.02742.x; PMID: 17581192
- Lavergne SN, Wang H, Callan HE, Park BK, Naisbitt DJ. “Danger” conditions increase sulfamethoxazole-protein adduct formation in human antigen-presenting cells. J Pharmacol Exp Ther 2009; 331:372 - 81; http://dx.doi.org/10.1124/jpet.109.155374; PMID: 19666748
- Sanderson JP, Naisbitt DJ, Farrell J, Ashby CA, Tucker MJ, Rieder MJ, et al. Sulfamethoxazole and its metabolite nitroso sulfamethoxazole stimulate dendritic cell costimulatory signaling. J Immunol 2007; 178:5533 - 42; PMID: 17442935
- Elsheikh A, Lavergne SN, Castrejon JL, Farrell J, Wang H, Sathish J, et al. Drug antigenicity, immunogenicity, and costimulatory signaling: evidence for formation of a functional antigen through immune cell metabolism. J Immunol 2010; 185:6448 - 60; http://dx.doi.org/10.4049/jimmunol.1000889; PMID: 20980635
- Martin AM, Almeida CA, Cameron P, Purcell AW, Nolan D, James I, et al. Immune responses to abacavir in antigen-presenting cells from hypersensitive patients. AIDS 2007; 21:1233 - 44; http://dx.doi.org/10.1097/QAD.0b013e3280119579; PMID: 17545699
- Kano Y, Shiohara T. The variable clinical picture of drug-induced hypersensitivity syndrome/drug rash with eosinophilia and systemic symptoms in relation to the eliciting drug. Immunol Allergy Clin North Am 2009; 29:481 - 501; http://dx.doi.org/10.1016/j.iac.2009.04.007; PMID: 19563993
- Hanafusa T, Azukizawa H, Matsumura S, Katayama I. The predominant drug-specific T-cell population may switch from cytotoxic T cells to regulatory T cells during the course of anticonvulsant-induced hypersensitivity. J Dermatol Sci 2012; 65:213 - 9; http://dx.doi.org/10.1016/j.jdermsci.2011.12.002; PMID: 22226608
- Illing PT, Vivian JP, Dudek NL, Kostenko L, Chen Z, Bharadwaj M, et al. Immune self-reactivity triggered by drug-modified HLA-peptide repertoire. Nature 2012; 486:554 - 8; PMID: 22722860
- Adam J, Eriksson KK, Schnyder B, Fontana S, Pichler WJ, Yerly D. Avidity determines T-cell reactivity in abacavir hypersensitivity. Eur J Immunol 2012; 42:1706 - 16; http://dx.doi.org/10.1002/eji.201142159; PMID: 22585534
- Chessman D, Kostenko L, Lethborg T, Purcell AW, Williamson NA, Chen Z, et al. Human leukocyte antigen class I-restricted activation of CD8+ T cells provides the immunogenetic basis of a systemic drug hypersensitivity. Immunity 2008; 28:822 - 32; http://dx.doi.org/10.1016/j.immuni.2008.04.020; PMID: 18549801
- Llano A, Brander C. Mechanisms involved in the Abacavir-mediated hypersensitivity syndrome. Cell Res 2012; http://dx.doi.org/10.1038/cr.2012.105; PMID: 22777424
- Pirmohamed M. Genetics and the potential for predictive tests in adverse drug reactions. Chem Immunol Allergy 2012; 97:18 - 31; http://dx.doi.org/10.1159/000335613; PMID: 22613851
- Bharadwaj M, Illing P, Theodossis A, Purcell AW, Rossjohn J, McCluskey J. Drug hypersensitivity and human leukocyte antigens of the major histocompatibility complex. Annu Rev Pharmacol Toxicol 2012; 52:401 - 31; http://dx.doi.org/10.1146/annurev-pharmtox-010611-134701; PMID: 22017685
- McCormack M, Alfirevic A, Bourgeois S, Farrell JJ, Kasperavičiūtė D, Carrington M, et al. HLA-A*3101 and carbamazepine-induced hypersensitivity reactions in Europeans. N Engl J Med 2011; 364:1134 - 43; http://dx.doi.org/10.1056/NEJMoa1013297; PMID: 21428769
- Ko TM, Chung WH, Wei CY, Shih HY, Chen JK, Lin CH, et al. Shared and restricted T-cell receptor use is crucial for carbamazepine-induced Stevens-Johnson syndrome. J Allergy Clin Immunol 2011; 128:1266 - 76, e11; http://dx.doi.org/10.1016/j.jaci.2011.08.013; PMID: 21924464
- Chung WH, Hung SI. Recent advances in the genetics and immunology of Stevens-Johnson syndrome and toxic epidermal necrosis. J Dermatol Sci 2012; 66:190 - 6; http://dx.doi.org/10.1016/j.jdermsci.2012.04.002; PMID: 22541332
- Viard I, Wehrli P, Bullani R, Schneider P, Holler N, Salomon D, et al. Inhibition of toxic epidermal necrolysis by blockade of CD95 with human intravenous immunoglobulin. Science 1998; 282:490 - 3; http://dx.doi.org/10.1126/science.282.5388.490; PMID: 9774279
- Nassif A, Bensussan A, Dorothée G, Mami-Chouaib F, Bachot N, Bagot M, et al. Drug specific cytotoxic T-cells in the skin lesions of a patient with toxic epidermal necrolysis. J Invest Dermatol 2002; 118:728 - 33; http://dx.doi.org/10.1046/j.1523-1747.2002.01622.x; PMID: 11918724
- Shiohara T, Kano Y. A complex interaction between drug allergy and viral infection. Clin Rev Allergy Immunol 2007; 33:124 - 33; http://dx.doi.org/10.1007/s12016-007-8010-9; PMID: 18094951
- Takahashi R, Kano Y, Yamazaki Y, Kimishima M, Mizukawa Y, Shiohara T. Defective regulatory T cells in patients with severe drug eruptions: timing of the dysfunction is associated with the pathological phenotype and outcome. J Immunol 2009; 182:8071 - 9; http://dx.doi.org/10.4049/jimmunol.0804002; PMID: 19494333
- Criado PR, Criado RF, Avancini Jde M, Santi CG. Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS) / Drug-induced Hypersensitivity Syndrome: a review of current concepts. An Bras Dermatol 2012; 87:435 - 49; http://dx.doi.org/10.1590/S0365-05962012000300013; PMID: 22714760
- Ju C, Reilly T. Role of immune reactions in drug-induced liver injury (DILI). Drug Metab Rev 2012; 44:107 - 15; http://dx.doi.org/10.3109/03602532.2011.645579; PMID: 22235834
- Tujios S, Fontana RJ. Mechanisms of drug-induced liver injury: from bedside to bench. Nat Rev Gastroenterol Hepatol 2011; 8:202 - 11; http://dx.doi.org/10.1038/nrgastro.2011.22; PMID: 21386809
- You Q, Cheng L, Reilly TP, Wegmann D, Ju C. Role of neutrophils in a mouse model of halothane-induced liver injury. Hepatology 2006; 44:1421 - 31; http://dx.doi.org/10.1002/hep.21425; PMID: 17133481
- Jenkins RE, Meng X, Elliott VL, Kitteringham NR, Pirmohamed M, Park BK. Characterisation of flucloxacillin and 5-hydroxymethyl flucloxacillin haptenated HSA in vitro and in vivo. Proteomics Clin Appl 2009; 3:720 - 9; http://dx.doi.org/10.1002/prca.200800222; PMID: 21136982
- Alfirevic A, Gonzalez-Galarza F, Bell C, Martinsson K, Platt V, Bretland G, et al. In silico analysis of HLA associations with drug-induced liver injury: use of a HLA-genotyped DNA archive from healthy volunteers. Genome Med 2012; 4:51; http://dx.doi.org/10.1186/gm350; PMID: 22732016
- Daly AK. Drug-induced liver injury: past, present and future. Pharmacogenomics 2010; 11:607 - 11; http://dx.doi.org/10.2217/pgs.10.24; PMID: 20415545
- Philp JR. Allergic Drug Reactions.; In: Walker HK, Hall WD, Hurst JW Ed. Clinical Methods: The History, Physical, and Laboratory Examinations. Boston, Butterworth Publishers 1990; Chapter 24.
- Bloemen K, Verstraelen S, Van Den Heuvel R, Witters H, Nelissen I, Schoeters G. The allergic cascade: review of the most important molecules in the asthmatic lung. Immunol Lett 2007; 113:6 - 18; http://dx.doi.org/10.1016/j.imlet.2007.07.010; PMID: 17765979
- Garratty G. Immune hemolytic anemia caused by drugs. Expert Opin Drug Saf 2012; 11:635 - 42; http://dx.doi.org/10.1517/14740338.2012.678832; PMID: 22502777
- Castells M, Sancho-Serra MD, Simarro M. Hypersensitivity to antineoplastic agents: mechanisms and treatment with rapid desensitization. Cancer Immunol Immunother 2012; http://dx.doi.org/10.1007/s00262-012-1273-x; PMID: 22576054
- Adam J, Pichler WJ, Yerly D. Delayed drug hypersensitivity: models of T-cell stimulation. Br J Clin Pharmacol 2011; 71:701 - 7; http://dx.doi.org/10.1111/j.1365-2125.2010.03764.x; PMID: 21480949
- Glimcher LH, Murphy KM. Lineage commitment in the immune system: the T helper lymphocyte grows up. Genes Dev 2000; 14:1693 - 711; PMID: 10898785
- Dong C. TH17 cells in development: an updated view of their molecular identity and genetic programming. Nat Rev Immunol 2008; 8:337 - 48; http://dx.doi.org/10.1038/nri2295; PMID: 18408735
- Hochrein H, O’Keeffe M. Dendritic cell subsets and toll-like receptors. Handb Exp Pharmacol 2008; 183:153 - 79; http://dx.doi.org/10.1007/978-3-540-72167-3_8; PMID: 18071659
- Nakanishi K. Basophils are potent antigen-presenting cells that selectively induce Th2 cells. Eur J Immunol 2010; 40:1836 - 42; http://dx.doi.org/10.1002/eji.201040588; PMID: 20518033
- Daubner B, Groux-Keller M, Hausmann OV, Kawabata T, Naisbitt DJ, Park BK, et al. Multiple drug hypersensitivity: normal Treg cell function but enhanced in vivo activation of drug-specific T cells. Allergy 2012; 67:58 - 66; http://dx.doi.org/10.1111/j.1398-9995.2011.02720.x; PMID: 21933197
- Muh HC, Tong JC, Tammi MT. AllerHunter: a SVM-pairwise system for assessment of allergenicity and allergic cross-reactivity in proteins. PLoS One 2009; 4:e5861; http://dx.doi.org/10.1371/journal.pone.0005861; PMID: 19516900
- Traidl-Hoffmann C, Jakob T, Behrendt H. Determinants of allergenicity. J Allergy Clin Immunol 2009; 123:558 - 66; http://dx.doi.org/10.1016/j.jaci.2008.12.003; PMID: 19152966
- Palikhe NS, Kim SH, Nam YH, Ye YM, Park HS. Polymorphisms of Aspirin-Metabolizing Enzymes CYP2C9, NAT2 and UGT1A6 in Aspirin-Intolerant Urticaria. Allergy Asthma Immunol Res 2011; 3:273 - 6; http://dx.doi.org/10.4168/aair.2011.3.4.273; PMID: 21966608
- Di Lorenzo G, Pacor ML, Candore G, Listi F, Ditta V, Leto-Barone MS, et al. Polymorphisms of cyclo-oxygenases and 5-lipo-oxygenase-activating protein are associated with chronic spontaneous urticaria and urinary leukotriene E4. Eur J Dermatol 2011; 21:47 - 52; PMID: 21227888
- Stevenson DD. Aspirin sensitivity and desensitization for asthma and sinusitis. Curr Allergy Asthma Rep 2009; 9:155 - 63; http://dx.doi.org/10.1007/s11882-009-0023-4; PMID: 19210906
- Rizk H. Role of aspirin desensitization in the management of chronic rhinosinusitis. Curr Opin Otolaryngol Head Neck Surg 2011; 19:210 - 7; http://dx.doi.org/10.1097/MOO.0b013e3283450102; PMID: 21372715
- Vasekar M, Craig TJ. ACE inhibitor-induced angioedema. Curr Allergy Asthma Rep 2012; 12:72 - 8; http://dx.doi.org/10.1007/s11882-011-0238-z; PMID: 22127615
- Torres MJ, Blanca M. The complex clinical picture of beta-lactam hypersensitivity: penicillins, cephalosporins, monobactams, carbapenems, and clavams. [xii.] Med Clin North Am 2010; 94:805 - 20, xii; http://dx.doi.org/10.1016/j.mcna.2010.04.006; PMID: 20609864
- Warrington R, Silviu-Dan F. Drug allergy. Allergy Asthma Clin Immunol 2011; 7:Suppl 1 S10; http://dx.doi.org/10.1186/1710-1492-7-S1-S10; PMID: 22165859
- Strom BL, Schinnar R, Apter AJ, Margolis DJ, Lautenbach E, Hennessy S, et al. Absence of cross-reactivity between sulfonamide antibiotics and sulfonamide nonantibiotics. N Engl J Med 2003; 349:1628 - 35; http://dx.doi.org/10.1056/NEJMoa022963; PMID: 14573734
- Zawodniak A, Lochmatter P, Beeler A, Pichler WJ. Cross-reactivity in drug hypersensitivity reactions to sulfasalazine and sulfamethoxazole. Int Arch Allergy Immunol 2010; 153:152 - 6; http://dx.doi.org/10.1159/000312632; PMID: 20413982
- Weberschock TB, Müller SM, Boehncke S, Boehncke WH. Tolerance to coxibs in patients with intolerance to non-steroidal anti-inflammatory drugs (NSAIDs): a systematic structured review of the literature. Arch Dermatol Res 2007; 299:169 - 75; http://dx.doi.org/10.1007/s00403-007-0757-6; PMID: 17492455
- Chaudhry T, Hissaria P, Wiese M, Heddle R, Kette F, Smith WB. Oral drug challenges in non-steroidal anti-inflammatory drug-induced urticaria, angioedema and anaphylaxis. Intern Med J 2012; 42:665 - 71; http://dx.doi.org/10.1111/j.1445-5994.2011.02601.x; PMID: 21981353
- Gallitelli M, Alzetta M. Icatibant: a novel approach to the treatment of angioedema related to the use of angiotensin-converting enzyme inhibitors. Am J Emerg Med 2011; http://dx.doi.org/10.1016/j.ajem.2011.09.014; PMID: 22100478
- Mertes PM, Demoly P, Malinovsky JM. Hypersensitivity reactions in the anesthesia setting/allergic reactions to anesthetics. Curr Opin Allergy Clin Immunol 2012; 12:361 - 8; http://dx.doi.org/10.1097/ACI.0b013e328355b82f; PMID: 22766618
- Liu A, Fanning L, Chong H, Fernandez J, Sloane D, Sancho-Serra M, et al. Desensitization regimens for drug allergy: state of the art in the 21st century. Clin Exp Allergy 2011; 41:1679 - 89; http://dx.doi.org/10.1111/j.1365-2222.2011.03825.x; PMID: 21883538
- Neuman MG, Cohen L, Nanau RM, Hwang PA. Genetic and immune predictors for hypersensitivity syndrome to antiepileptic drugs. Transl Res 2012; 159:397 - 406; http://dx.doi.org/10.1016/j.trsl.2012.01.004; PMID: 22500513
- Daly AK, Day CP. Genetic association studies in drug-induced liver injury. Drug Metab Rev 2012; 44:116 - 26; http://dx.doi.org/10.3109/03602532.2011.605790; PMID: 21913872
- Cernadas JR, Brockow K, Romano A, Aberer W, Torres MJ, Bircher A, et al, European Network of Drug Allergy and the EAACI interest group on drug hypersensitivity. General considerations on rapid desensitization for drug hypersensitivity - a consensus statement. Allergy 2010; 65:1357 - 66; http://dx.doi.org/10.1111/j.1398-9995.2010.02441.x; PMID: 20716314
- de Groot H, Mulder WM. Clinical practice: drug desensitization in children. Eur J Pediatr 2010; 169:1305 - 9; http://dx.doi.org/10.1007/s00431-010-1236-1; PMID: 20571825
- Woo HY, Kim YS, Kang NI, Chung WC, Song CH, Choi IW, et al. Mechanism for acute oral desensitization to antibiotics. Allergy 2006; 61:954 - 8; http://dx.doi.org/10.1111/j.1398-9995.2006.01147.x; PMID: 16867049
- Park JS, Choi IH, Lee DG, Han SS, Ha TY, Lee JH, et al. Anti-IL-4 monoclonal antibody prevents antibiotics-induced active fatal anaphylaxis. J Immunol 1997; 158:5002 - 6; PMID: 9144520
- del Carmen Sancho M, Breslow R, Sloane D, Castells M. Desensitization for hypersensitivity reactions to medications. Chem Immunol Allergy 2012; 97:217 - 33; http://dx.doi.org/10.1159/000335637; PMID: 22613865
- MacGlashan DJ Jr.. Subthreshold desensitization of human basophils re-capitulates the loss of Syk and FcεRI expression characterized by other methods of desensitization. Clin Exp Allergy 2012; 42:1060 - 70; http://dx.doi.org/10.1111/j.1365-2222.2012.04013.x; PMID: 22702505
- De Weck AL, Jeunet F, Schulz KH, Louis P, Girard JP, Grilliat JP, et al. Clinical trial of Ro 6-0787, a monovalent specific hapten inhibitor of penicillin allergy. Z Immunitatsforsch Exp Klin Immunol 1975; 150:138 - 60; PMID: 127471
- Stevenson DD, Simon RA, Mathison DA. Aspirin-sensitive asthma: tolerance to aspirin after positive oral aspirin challenges. J Allergy Clin Immunol 1980; 66:82 - 8; http://dx.doi.org/10.1016/0091-6749(80)90143-8; PMID: 7381126
- Stevenson DD, Pleskow WW, Simon RA, Mathison DA, Lumry WR, Schatz M, et al. Aspirin-sensitive rhinosinusitis asthma: a double-blind crossover study of treatment with aspirin. J Allergy Clin Immunol 1984; 73:500 - 7; http://dx.doi.org/10.1016/0091-6749(84)90361-0; PMID: 6368649
- Sweet JM, Stevenson DD, Simon RA, Mathison DA. Long-term effects of aspirin desensitization--treatment for aspirin-sensitive rhinosinusitis-asthma. J Allergy Clin Immunol 1990; 85:59 - 65; http://dx.doi.org/10.1016/0091-6749(90)90222-P; PMID: 2299107
- Berges-Gimeno MP, Simon RA, Stevenson DD. Long-term treatment with aspirin desensitization in asthmatic patients with aspirin-exacerbated respiratory disease. J Allergy Clin Immunol 2003; 111:180 - 6; http://dx.doi.org/10.1067/mai.2003.7; PMID: 12532116
- Stevenson DD, Hankammer MA, Mathison DA, Christiansen SC, Simon RA. Aspirin desensitization treatment of aspirin-sensitive patients with rhinosinusitis-asthma: long-term outcomes. J Allergy Clin Immunol 1996; 98:751 - 8; http://dx.doi.org/10.1016/S0091-6749(96)70123-9; PMID: 8876550
- Parikh AA, Scadding GK. Intranasal lysine-aspirin in aspirin-sensitive nasal polyposis: a controlled trial. Laryngoscope 2005; 115:1385 - 90; http://dx.doi.org/10.1097/01.MLG.0000166702.38850.1B; PMID: 16094110
- Ogata N, Darby Y, Scadding G. Intranasal lysine-aspirin administration decreases polyp volume in patients with aspirin-intolerant asthma. J Laryngol Otol 2007; 121:1156 - 60; http://dx.doi.org/10.1017/S0022215107000515; PMID: 17697437
- Cianferoni A, Schroeder JT, Kim J, Schmidt JW, Lichtenstein LM, Georas SN, et al. Selective inhibition of interleukin-4 gene expression in human T cells by aspirin. Blood 2001; 97:1742 - 9; http://dx.doi.org/10.1182/blood.V97.6.1742; PMID: 11238116
- Turcanu V, Maleki SJ, Lack G. Characterization of lymphocyte responses to peanuts in normal children, peanut-allergic children, and allergic children who acquired tolerance to peanuts. J Clin Invest 2003; 111:1065 - 72; PMID: 12671056
- Tsuge I, Okumura A, Kondo Y, Itomi S, Kakami M, Kawamura M, et al. Allergen-specific T-cell response in patients with phenytoin hypersensitivity; simultaneous analysis of proliferation and cytokine production by carboxyfluorescein succinimidyl ester (CFSE) dilution assay. Allergol Int 2007; 56:149 - 55; http://dx.doi.org/10.2332/allergolint.O-06-457; PMID: 17460442
- Okumura A, Tsuge I, Kubota T, Kurahashi H, Natsume J, Negoro T, et al. Phenytoin desensitization monitored by antigen specific T cell response using carboxyfluorescein succinimidyl ester dilution assay. Eur J Paediatr Neurol 2007; 11:385 - 8; http://dx.doi.org/10.1016/j.ejpn.2007.03.001; PMID: 17428709
- Mizukawa Y, Yamazaki Y, Shiohara T. In vivo dynamics of intraepidermal CD8+ T cells and CD4+ T cells during the evolution of fixed drug eruption. Br J Dermatol 2008; 158:1230 - 8; http://dx.doi.org/10.1111/j.1365-2133.2008.08516.x; PMID: 18363767
- Mizukawa Y, Shiohara T. Fixed drug eruption: a prototypic disorder mediated by effector memory T cells. Curr Allergy Asthma Rep 2009; 9:71 - 7; http://dx.doi.org/10.1007/s11882-009-0011-8; PMID: 19063828
- Teraki Y, Shiohara T. Successful desensitization to fixed drug eruption: the presence of CD25+CD4+ T cells in the epidermis of fixed drug eruption lesions may be involved in the induction of desensitization. Dermatology 2004; 209:29 - 32; http://dx.doi.org/10.1159/000078583; PMID: 15237264
- Lin D, Li WK, Rieder MJ. Cotrimoxazole for prophylaxis or treatment of opportunistic infections of HIV/AIDS in patients with previous history of hypersensitivity to cotrimoxazole. Cochrane Database Syst Rev 2007; CD005646; PMID: 17443608
- Fam AG, Dunne SM, Iazzetta J, Paton TW. Efficacy and safety of desensitization to allopurinol following cutaneous reactions. Arthritis Rheum 2001; 44:231 - 8; http://dx.doi.org/10.1002/1529-0131(200101)44:1<231::AID-ANR30>3.0.CO;2-7; PMID: 11212165
- Demoly P, Messaad D, Sahla H, Fabre J, Faucherre V, André P, et al. Six-hour trimethoprim-sulfamethoxazole-graded challenge in HIV-infected patients. J Allergy Clin Immunol 1998; 102:1033 - 6; http://dx.doi.org/10.1016/S0091-6749(98)70343-4; PMID: 9847446
- Cernadas JR, Brockow K, Romano A, Aberer W, Torres MJ, Bircher A, et al, European Network of Drug Allergy and the EAACI interest group on drug hypersensitivity. General considerations on rapid desensitization for drug hypersensitivity - a consensus statement. Allergy 2010; 65:1357 - 66; http://dx.doi.org/10.1111/j.1398-9995.2010.02441.x; PMID: 20716314
- Vultaggio A, Maggi E, Matucci A. Immediate adverse reactions to biologicals: from pathogenic mechanisms to prophylactic management. Curr Opin Allergy Clin Immunol 2011; 11:262 - 8; http://dx.doi.org/10.1097/ACI.0b013e3283464bcd; PMID: 21460715
- Yoshizawa S, Yasuoka A, Kikuchi Y, Honda M, Gatanaga H, Tachikawa N, et al. A 5-day course of oral desensitization to trimethoprim/sulfamethoxazole (T/S) in patients with human immunodeficiency virus type-1 infection who were previously intolerant to T/S. Ann Allergy Asthma Immunol 2000; 85:241 - 4; http://dx.doi.org/10.1016/S1081-1206(10)62474-X; PMID: 11030281