Abstract
A DNA vaccination approach was used in the current study to screen for the immunogenicity of different fragments of toxin A and toxin B from Clostridium difficile. With this approach, protein antigens do not need to be produced in vitro and the immunogenicity of candidate C. difficile antigens can be identified directly in animals. Codon optimized toxin gene fragments were individually cloned into the DNA vaccine vector and tested in mice and rabbits for their ability to elicit C. difficile toxin-specific antibody responses. Only a subset of the C. difficile toxin fragments, including the C-terminal receptor binding domain of toxin A and a novel N-terminal enzymatic domain of toxin B, were able to elicit protective antibody responses as determined by protection of target cells in a cytotoxicity assay or by preventing death of mice in a passive antibody protection study. Significantly, antibodies elicited by the novel N-terminus of the toxin B DNA vaccine were able to increase the level of protection when used in combination with anti-toxin A antibodies in a toxin challenge model in mice.
Introduction
Clostridium difficile is an anaerobic Gram-positive bacillus that possesses the ability to form spores resistant to many commonly used hospital disinfectants and can survive on medical devices, floors and on the hands of medical staff for several months.Citation1-Citation3C. difficile-associated diarrhea is one of the most significant emerging hospital acquired infections in Western countries and its prevalence has increased at an alarming rate over the past two decades. Less developed countries such as China have also seen C. difficile infection in specific hospital patient populations.Citation4
C. difficile produces two key virulence determinants, toxin A (TcdA) and toxin B (TcdB). The relative importance of these two toxins has been re-examined in recent studies. Earlier literature indicates that toxin A alone is sufficient to elicit the symptoms of C. difficile infection while toxin B is not able to do so unless it is mixed with toxin A or there is gut mucosa damage.Citation5 However, one recent study suggested that toxin B is essential and a strain producing toxin A alone is avirulent.Citation6 However, this finding was not held long before a new report showed that isogenic mutants of C. difficile producing either toxin A or toxin B alone could cause fulminant disease in a hamster model.Citation7 More significantly, by using a gene knockout system to inactivate the toxin genes, this study also demonstrated that C. difficile strains producing either one or both toxins showed cytotoxic activities in vitro and virulence in vivo, thus re-establishing the significance of both toxins.
These studies provide important guidance on the development of preventive and therapeutic approaches in controlling C. difficile infection. Because the widespread use of broad-spectrum antibiotics may be a key factor in causing C. difficile infection, choices for C. difficile treatment using conventional antibiotics is limited. The rate of treatment failure is high with existing antibiotic treatments and recurrent infections are frequently observed.Citation8-Citation14 Therefore, there is a great need to develop passive antibody therapies or active immunization with vaccines targeting key virulence factors.Citation15
Toxin A and toxin B are two key targets for vaccines or passive antibody therapies against C. difficile. Due to the large size of both toxins, such efforts usually only use part of each toxin sequence as the antigens. Previous studies have mainly identified the C-terminal domain for both toxin A and toxin B,Citation16 toxin A C-terminal repeating unitsCitation17,Citation18 or even one or multiple copies of antigenic peptides in this region as the main antigenic regions that can elicit protective antibody responses.Citation19,Citation20 In addition to protein- or subunit-based vaccines, novel bacterial vectors have also been used to successfully deliver antigenic domains to elicit protective immunity.Citation21-Citation23 One of the advantages of these vectors is that they can be delivered through mucosal routes in animal studies to induce both systemic and mucosal immune responses.
Among all of the aforementioned studies, the C-terminal receptor binding domain of toxin A has been the main antigen. Recently, Gardiner et al. reported that a DNA vaccine expressing the same C-terminal receptor binding domain of TcdA induced humoral protective immunity in miceCitation24 and Seregin et al. demonstrated that the C-terminal domain expressed by a human type 5 adenovirus (Ad5) vector could induce cell-mediated immune responses and lead to protection against TcdA challenge in mice.Citation25 There are fewer reports on vaccine development based on toxin B and there is even less information on whether other regions of C. difficile toxins can also serve as protective antigens. With recent findings that toxin B is essential for virulence of C. difficileCitation6 and the fact that naturally occurring A-B+ strains exist and may cause clinical C. difficile infection,Citation26,Citation27 it is now important to identify novel region(s) on toxin B antigens that can maximize the protective efficacy of C. difficile vaccines. In the current study, we used the DNA vaccination approach as a tool to screen the immunogenicity of various toxin A and toxin B fragments. The findings from this study point to the potential of the N-terminus of toxin B as a candidate for the development of vaccines or antibody-based therapeutics against C. difficile.
Results
Toxin A and toxin B DNA vaccine design
In order to achieve high level expression of candidate C. difficile toxin antigens in mammalian cells, as would be required for DNA vaccines, codons from the original C. difficile toxins gene sequences were modified to increase the usage of codons preferred by mammalian cells. At the same time, it will be desirable to accommodate the potential production of recombinant C. difficile toxin proteins in a bacterial expression system, such as E. coli, for protein-based C. difficile vaccine development. Therefore, we have produced codon optimized C. difficile toxin gene sequences that have the potential to achieve high level expression in both mammalian and E. coli systems as previously described.Citation28
Based on function, toxin A can be divided into four major domains: the N-terminal enzymatic (catalytic) domain, the cysteine protease domain, the translocation domain and the C-terminal receptor binding domain (RBD).Citation29 Using a codon optimized C. difficile toxin A gene sequence as the template, five toxin A DNA vaccine inserts were produced and cloned into DNA vaccine vector, pJW4303 (). The first toxin A DNA vaccine insert, TcdA-N, codes for the 544 aa of the entire enzymatic domain. The second insert, TcdA-T1, codes for the next 929 aa, which falls in the N-terminal side of the translocation domain, and the third insert, TcdA-T2, codes for the 915 aa spanning the remaining part of translocation domain and part of RBD. The last two toxin A DNA inserts (TcdA-C and tPA-TcdA-C) express the same 324 aa region of the C-terminal part of RBD. The only difference between these two inserts is that the tPA-TcdA-C has an extra leader sequence from the human tissue plasminogen activator (tPA).
Figure 1. Schematic designs of DNA vaccines expressing various regions of C. difficile toxins A and B. (A) C. difficile toxin A (TcdA) DNA vaccines; (B) C. difficile toxin B (TcdB) DNA vaccines. The amino acid positions for corresponding protein segments are indicated.
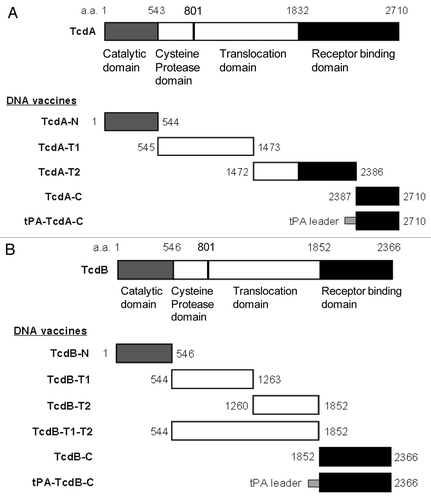
Similar to toxin A, toxin B can be divided into three major functional domains: the N-terminal glucosyltransferase domain; the middle translocation domain; and the C-terminal RBD. Using the codon optimized C. difficile toxin B gene sequence as the template, a total of six toxin B DNA vaccine inserts were produced in the current study to test the immunogenicity of different regions of the toxin B protein (). The first toxin B DNA vaccine insert, TcdB-N, codes for the 546 aa of the entire enzymatic domain. The next two inserts, TcdB-T1 (718 aa) and TcdB-T2 (593aa), cover the entire translocation domain. A longer version of the DNA vaccine insert, TcdB-T1-T2, is a combination of TcdB-T1 and TcdB-T2. The last two toxin B DNA inserts (TcdB-C and tPA-TcdB-C) express the same 515 aa of the C-terminal RBD. The only difference between these two inserts is that the tPA-TcdB-C has an extra tPA leader sequence.
The above toxin A and toxin B DNA vaccine inserts were individually cloned into the DNA vaccine vector pJW4303 and then verified by restriction enzyme digestion and DNA sequencing before testing their immunogenicity in both mouse and rabbit models.
Immunogenicity of TcdA and TcdB DNA vaccines
Mouse immunogenicity studies were conducted as a screening step to identify the overall ability of the DNA vaccines to elicit antibody responses against the autologous regions from toxin A and toxin B antigens. Each group of mice was immunized by one toxin A or toxin B DNA vaccine.
No antigen-specific serum antibody responses were detected in mice immunized with TcdA-N, TcdA-T1 or TcdA-T2 DNA vaccines based on ELISA or Western blot analysis (data not shown). One extra group of mice was immunized with the combination of TcdA-T1 and TcdA-T2 DNA vaccines and again no detectable TcdA-T antigen-specific antibodies were identified.
In contrast, immune sera from both TcdA-C and tPA-TcdA-C groups showed positive antibody responses. When the kinetics of mouse antibody responses was analyzed, two out of seven mice in the tPA-TcdA-C DNA vaccine group had detectable antigen-specific antibody responses after only one immunization while none of the mice in the TcdA-C DNA group showed any antibody responses after one immunization. After two immunizations, all seven mice in the tPA-TcdA-C DNA vaccine group had positive antibody responses but only five of seven mice in the TcdA-C DNA vaccine group had positive antibody responses. After three immunizations, all animals in both groups showed positive antibody responses and the antibody responses were further boosted to very high levels after the fourth immunization (). End-titration titers for both groups were very high (~1:106) at week 10 (two weeks after the fourth and last immunization); there were no significant differences in the end titration antibody titers between the two groups (). The control group, in which animals received empty DNA vector, did not have any significant antibody responses against the TcdA-C antigen ().
Figure 2. Antibody responses elicited by C. difficile toxin A DNA vaccines as measured by ELISA. (A) Temporal TcdA-C-specific antibody responses in mouse sera (1:500 dilution) receiving empty DNA vector pJW4303, TcdA-C DNA and tPA-TcdA-C DNA, respectively. Each curve represents the mean OD values plus standard deviation of antibody response for each mouse group. The arrows indicate the time points of DNA immunizations. (B) Peak level TcdA-C-specific IgG titers in individual mouse sera at two weeks after the 4th DNA immunization for different mouse groups. Each dot represents one animal. (C) Peak level TcdA-C-specific serum IgG titers in rabbits at two weeks after the 4th DNA immunization. Each dot represents one rabbit.
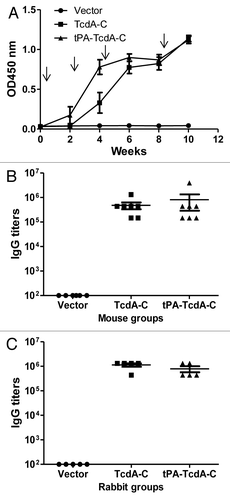
New Zealand White (NZW) rabbits were immunized with the same pairs of TcdA-C DNA vaccines to produce large quantity of hyperimmune sera for both in vitro and in vivo protection studies (see below). Again, peak level TcdA-C-specific antibody titers were very similar between animals receiving TcdA-C and tPA-TcdA-C DNA vaccines while the pJW4303 vector group did not show antigen-specific antibody responses ().
For toxin B DNA vaccines, no antigen-specific serum antibody responses were detected in mice immunized with TcdB-T1, TcdB-T2 or TcdB-T1-T2 DNA vaccines based on ELISA or Western blot analysis (data not shown). Different from toxin A DNA vaccines, both C-terminal toxin B DNA vaccines (TcdB-C and tPA-TcdB-C) failed to elicit specific antibodies against the autologous toxin B antigen segment (data not shown).
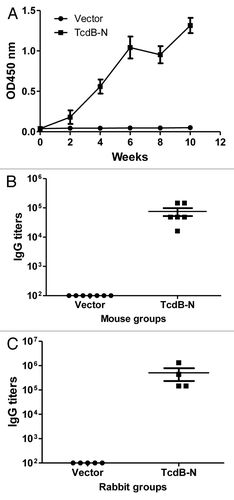
In contrast, mouse immune sera from the TcdB-N DNA vaccine group showed positive antibody responses. After two immunizations, all animals in the same group developed antibody responses against the TcdB-N antigen while there were no specific antibody responses in the control group (empty DNA vaccine vector pJW4303) (). Titers went up further with the third and fourth immunizations. Peak level antibody titers in the TcdB-N vaccine group reached about 1:105 (). Similarly, high titer anti-TcdB-N antibody responses were elicited in rabbits that received the same TcdB-N DNA vaccine ().
Figure 3. TcdB-N-specific IgG responses measured by ELISA. (A) Temporal TcdB-N-specific antibody responses in mouse sera (at 1:500 dilution) immunized with either the vector or TcdB-N DNA vaccine. Each curve represents the mean OD values plus standard deviation for each mouse group. The arrows indicate the time points of DNA immunizations. (B and C) Peak level serum TcdB-N-specific IgG titers in individual mice (B) and rabbits (C), both at two weeks after the 4th DNA immunization, respectively. Each dot represents one animal.
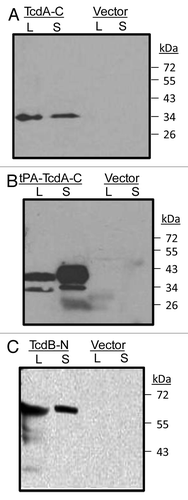
The specificity of animal immune sera was confirmed by Western blot analysis. Rabbit sera immunized with either TcdA-C () or tPA-TcdA-C () DNA vaccines were able to recognize TcdA-C antigen in both cell lysate and supernatant of 293T cell cultures that were transfected with one of these two DNA vaccines. Similarly, rabbit sera immunized with TcdB-N DNA vaccine recognized the TcdB-N antigen in both cell lysate and supernatant of 293T cell cultures ().
Figure 4. Western blot analysis of rabbit immune serum against toxin A C-terminal domains expressed in 293T cells (both supernatant, S and cell lysate, L) from either (A) TcdA-C or (B) tPA-TcdA-C DNA vaccines. DNA vector pJW4303 was used as a negative control. Serum from one TcdA-C DNA immunized rabbit (at 1:500 dilution) was used as the detection antibody. (C) western blot analysis of antigen expression from TcdB-N DNA vaccine transfected 293T cells. Cells transfected with the empty vector were used as negative controls. Serum from one TcdB-N-immunized rabbit (at 1:500 dilution) was used as the detection antibody.
Protective potential of rabbit immune sera elicited by either TcdA-C or TcdB-N DNA vaccines
Rabbit immune sera were further analyzed for their ability to protect cells and animals from toxin challenge. Concentrated bacteria culture filtrate was prepared and the presence and amounts of toxin A and toxin B in such preparations were determined by ELISA using commercial reagents.
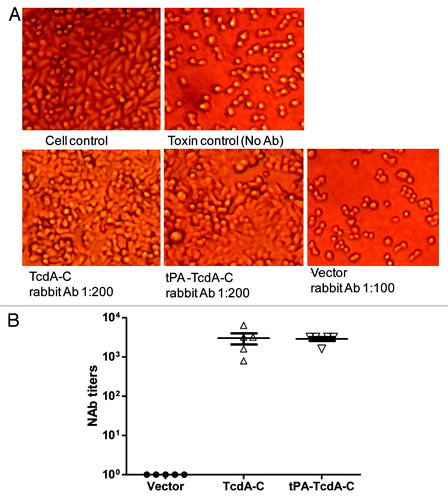
Rabbit immune sera elicited by TcdA-C or tPA-TcdA-C DNA vaccines were protective against toxin A in HT-29 cells. As shown in , at 1:200 dilution, rabbit sera from either TcdA-C or tPA-TcdA-C vaccine groups protected cells from cytotoxic activity while the more concentrated vector control rabbit sera, at 1:100 dilution, failed to protect cells (). The final neutralizing antibody titers for sera from both groups were very similar, both at ~1:5,000 ().
Figure 5. Anti-TcdA neutralizing activity measured against HT-29 cells. (A) Light microscopic images of HT-29 cells with various treatments: “Cell control” lacks treatment by both diluted bacteria culture filtrate and rabbit immune sera; “Toxin control” is the addition of diluted bacteria culture filtrate without any rabbit immune sera; “TcdA-C,” “tPA-TcdA-C” or “Vector” indicates that the cells were cultured with diluted bacteria culture filtrate pre-incubated with sera from one of the following rabbit groups: TcdA-C, tPA-TcdA-C or empty vector. Sera were diluted at 1:200 except for vector control group sera, which was diluted at 1:100 dilution. (B) Summary of anti-TcdA neutralizing antibody titers (NAb) in individual rabbit sera two weeks after the 4th DNA immunization.
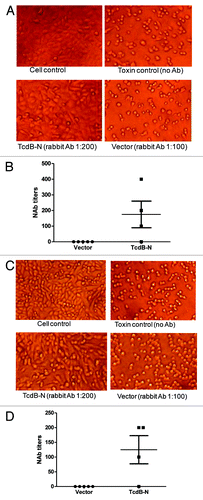
Rabbit immune sera elicited by the TcdB-N DNA vaccine were tested for protective antibody activities against toxin B in Vero and CHO cells. TcdB-N immunized rabbit sera (at 1:200 dilution) protected Vero cells from cytotoxic activity while the more concentrated vector control rabbit serum at 1:100 dilution failed to protect the cells (). The mean neutralizing antibody titer against toxin B was ~1:200 (). Similar protective activities were shown in CHO cells () except the mean neutralizing antibody titer was lower at ~1:125 ().
Figure 6. Anti-TcdB neutralizing activities measured in CHO (A and B) or Vero (C and D) cells. (A and C) Examples of CHO (A) and Vero cells (C), respectively. “Cell control,” without addition of both diluted bacteria culture filtrate and antibody; “Toxin control,” with addition of bacteria culture filtrate but without rabbit serum treatment; “TcdB-N” or “Vector” indicate that the cells were cultured with bacteria culture filtrate pre-incubated with TcdB-N or empty vector immunized rabbit serum, respectively. (B and D) Anti-TcdB neutralizing antibody titers in CHO (B) and Vero cells (D), respectively. Individual rabbit sera were measured at two weeks after the 4th DNA immunization in each DNA immunization group: empty DNA vector, and TcdB-N.
Two in vivo passive antibody protection studies were conducted in BALB/c mice. In one study, mice were challenged with the commercial C. difficile toxin A product (). Animals that received either no treatment or pre-bleed rabbit sera all died within 24–36 h after challenge, while 100% of animals who received rabbit immune sera elicited by the TcdA-C DNA vaccine survived the challenge ().
Figure 7. Passive antibody protection of mice against commercial toxin A product (A) or C. difficile concentrated culture filtrates (B and C). The filtrate was incubated with various rabbit immune sera and prebleed sera, as indicated. *Indicates p < 0.05; **indicates p < 0.01, when compared with the group inoculated with pre-bleed. See “Materials and Methods” section for study group designs.
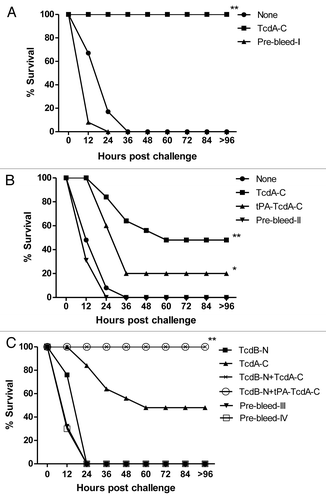
In the second study, concentrated C. difficile bacterial culture filtrate was used as the challenge (). Again, all the animals in the no-treatment and pre-bleed treatment groups died quickly after the challenge. Rabbit immune sera elicited by the two TcdA-C DNA vaccines could partially protect the animals: 20% of animals survived in the group receiving the tPA-TcdA-C DNA vaccine rabbit serum and 50% survived in the group receiving the TcdA-C DNA vaccine rabbit serum. Both toxin A and toxin B were confirmed in VPI 10463 culture filtrate via ELISA and therefore, anti-toxin A antibodies alone may not be able to provide full protection.
An in vivo passive antibody protection study was then conducted in mice to test the protective potential of rabbit sera elicited by the TcdB-N DNA vaccine against concentrated C. difficile bacterial culture filtrate (). All of the animals in the pre-bleed treatment group died within 24 h after challenge. When used alone, rabbit immune sera elicited by the TcdB-N DNA vaccine also failed to protect the animals. When TcdB-N rabbit immune sera were used in combination with either TcdA-C or tPA-TcdA-C rabbit immune sera, 100% of the animals survived although TcdA-C rabbit immune sera alone could only achieve 50% protection. By blocking the effect of toxin B, anti-toxin B antibodies provided the additional protective effect when used in combination with anti-toxin A antibodies; however, anti-toxin B antibodies alone were not sufficient to demonstrate even partial protection when the effect of toxin A was not blocked.
Discussion
Currently there is an active effort by several major pharmaceutical companies toward the development of vaccines against C. difficile infection. Passive protection with anti-toxin monoclonal antibodies has also been proven effective in reducing recurrent C. difficile infection.Citation30 The key element for both active and passive vaccination approaches is the discovery of high quality protective antibody responses against two key C. difficile toxins.Citation24 Traditionally, anti-toxin A antibodies have been widely studied but less is known about the immunogenicity potential of toxin B. The size and the highly unstable nature of both toxins, especially toxin B, have made the use of full-length recombinant protein-based vaccines less practical. While it is generally known that the C-terminal receptor binding regions of both toxins A and B are ideal candidates for eliciting protective antibody responses, it is not known whether other areas of both toxins can elicit protective antibody responses.
The current study focused on the immunogenicity of different domains of both toxins. A key finding from our study is that the N-terminal region of toxin B protein can serve as an excellent immunogen to elicit protective antibodies, effective not only in protecting cells in an in vitro cytotoxicity assay but also functional in improving the protection of mice against a lethal C. difficile toxin challenge when used in combination with anti-toxin A antibodies. The discovery of this novel N-terminal domain of TcdB as a protective immunogen will offer more options to design the next generation subunit-based TcdB or TcdA-TcdB combination vaccines.
One previous study reported that monoclonal antibody 103–174, which binds to aa 1–592 at the N-terminal end of toxin B, showed neutralizing activity against toxin B in a MR-90 cell line-based cytotoxicity assay but failed to protect lethal challenge in a mouse model against toxin B challenge.Citation31 Our data provided further confirmation to support the idea that the N-terminal region of toxin B can serve as a protective immunogen. In light of our data, anti-toxin B antibodies appear insufficient to provide full protection. However, when combined with anti-toxin A antibodies, they are fully protective. This finding suggests that the N-terminal domain of toxin B should also be considered as a candidate immunogen in C. difficile vaccine design. This region can also be used to elicit antibodies for therapeutic applications as a treatment to control C. difficile infection.
Other results included in the current report are either expected or do not reveal any new information. Two toxin A antigen designs (both targeting the C-terminal RBD region) were able to elicit high titer antibodies that were protective as shown by both in vitro and in vivo toxin challenge studies. Other antigen designs failed to elicit antigen-specific antibodies reflecting either their overall poor immunogenicity (such as the hydrophobic region in the middle of toxin B) or the low level antigen expression by two C-terminal toxin B DNA vaccines. No antigen-specific serum antibody responses were detected in mice immunized with TcdA-N, TcdA-T1 or TcdA-T2 DNA vaccines based on ELISA or Western blot analysis, which may be due to the low protein expression levels by the current DNA vaccine constructs or low immunogenicity of such domains. Such negative data also provides useful information when other vaccination approaches are tested.
Our study also demonstrates that adding a tPA leader sequence may increase the rate of early detection of antigen-specific binding antibodies following vaccination with C-terminal toxin A DNA vaccines but at peak levels, the difference was less obvious. There was no difference in protective antibody responses elicited by these two TcdA-C DNA vaccines when measured by an in vitro cytotoxicity assay. Interestingly, in the mouse challenge study, TcdA-C sera provided better protection (50%) than tPA-TcdA-C sera (20%), which may imply that the quality of antibodies elicited by the natural C-terminal domain of toxin A, without any extra leader sequence, are more functionally relevant.
It is important to note that we did not use a real vaccination study model to directly test the protection against C. difficile disease by DNA vaccines. Both in vitro and in vivo studies were conducted to demonstrate the protective nature of positive antibody responses elicited by TcdA-C and TcdB-N DNA vaccines; however, these studies do not directly demonstrate the protection efficacy of C. difficile DNA vaccines. Identification of protective antibodies against toxins in our study will be a necessary first step to confirm the immunogenicity of candidate toxin antigenic domains, especially for the N-terminal domain of the toxin B protein.
DNA vaccination offers a unique technology platform to study the optimal antigenic regions from both toxins as this approach is able to test the immunogenicity of candidate antigens in animals directly without first producing actual antigenic proteins in vitro. Once high level antibody responses are elicited, the same antigen region can be used to produce subunit-based recombinant toxin proteins as vaccines. Furthermore, the same toxin antigens can be used to produce hyperimmune sera that can be administered for passive antibody protection. Monoclonal antibodies can also be generated from a high responder host (animal or human volunteers) who received the novel N-terminal region from the C. difficile toxin B.
It is also possible that immunogens expressed in vivo have a better chance to maintain antigen conformation compared with inactivated toxoid antigens as previously demonstrated in HIV vaccine studies.Citation32 This may be the reason why our TcdB-N DNA vaccine was able to elicit protective antibody responses while no previous immunization studies have reported similar results. Future studies will need to compare the immunogenicity between DNA and recombinant subunit protein vaccines targeting the same N-terminal region of toxin B to demonstrate the relative effectiveness between these two types of vaccines.
Our data provide interesting insight related to the relative roles of two toxins in C. difficile infection. While the traditional view held that toxin A alone was able to evoke the symptoms of C. difficile infection,Citation5 more recent findings indicate that toxin B is essential for virulence of C. difficileCitation6 or, at least, that both toxin A and toxin B are important.Citation7 Our work indicates that the biological outcome of a purified toxin challenge is different from a challenge by C. difficile culture filtrate, which more closely emulates natural infection. It may be easier to protect against challenge by purified toxin A but much more difficult to protect against challenge by natural C. difficile culture filtrate as it contains both toxin A and toxin B components at an unknown ratio. At the same time, the recombinant toxin B used in this study is not a very effective challenge, making protection studies with anti-toxin B antibodies difficult. Only when the role of anti-toxin B antibodies are measured in combination with anti-toxin A antibodies against toxin A and toxin B in C. difficile culture filtrate challenge and the efficacy of anti-toxin B antibodies will be better demonstrated.
Results from this study act to facilitate or confirm the immunogen design of C. difficile vaccines using selected domains from both toxins. Such subunit antigen-based vaccines can be in the form of DNA vaccines,Citation24 as shown here, or alternatively, recombinant protein-based vaccines or a vector-based vaccine approach.Citation25 Furthermore, a prime-boost approach, to combine two different vaccine modalities to sequentially deliver the same immunogens, may further enhance the levels of protective antibody responses as demonstrated in other non-C. difficile vaccine studies.Citation33-Citation35
Materials and Methods
Codon optimization of C. difficile toxin genes
The codon usages of tcdA (gene coding for the toxin A of C. difficile) and tcdB (gene coding for the toxin B of C. difficile) (GenBank accession numbers: M30307 and X53138, respectively) were analyzed with the MacVector software 7.2 against codon preference of Homo sapiens and Escherichia coli. While the new codon optimized tcdA and tcdB genes were designed mainly for the development of TcdA and TcdB DNA vaccines, the less optimal codons in the tcdA and tcdB genes were changed to the preferred codons to allow a higher expression of the resulting TcdA and TcdB protein domains in both mammalian and E. coli expression systems.Citation28 Sequence optimization was also performed to make the mRNA more stable and to make the gene more favorable for transcriptional and translational processes. During sequence optimization, the following cis-acting sequence motifs were avoided: internal TATA-boxes, chi-sites and ribosomal entry sites; AT-rich or GC-rich sequence stretches; INS (inhibitory sequences), CRS (cis-acting repressor sequences) elements; cryptic splice donor and acceptor sites; and branch points. The codon optimized tcdA and tcdB gene sequences were chemically synthesized at GeneArt (Regensburg, Germany) and restriction enzyme sites for subcloning purposes were added immediately upstream of the start codon and downstream of the stop codon.
Design and cloning of C. difficile toxin DNA vaccines
The codon optimized gene fragments of toxin A TcdA-N, TcdA-T1, TcdA-T2 and TcdA-C were chemically synthesized by GeneArt. Restriction enzyme sites of PstI for TcdA-C and HindIII for TcdA-N, TcdA-T1 and TcdA-T2 were included immediately upstream of the start codon. Restriction enzyme site BamHI was included downstream of the stop codon for all inserts. These inserts were individually subcloned into the DNA vaccine vector pJW4303. For pJW4303-tPA-TcdA-C, which had the tissue plasminogen activator (tPA) signal peptide sequence upstream of TcdA-C, a tPA-TcdA-C insert was first produced by PCR from the TcdA-C insert using the primers F1 (5′ GTCACTTCGCTAGCGCCAGCACCGGCTACACCAGC 3′), incorporating a NheI restriction site, and primer R1 (5′ AACTTCGGATCCTCATCAGCCGTAGATG 3′), incorporating a BamHI restriction site, and then subcloned into pJW4303.
The codon optimized fragments of toxin B TcdB-N, TcdB-T1, TcdB-T2 and TcdB-C were also chemically synthesized by GeneArt with HindIII site added immediately upstream of the start codon and BamHI site located downstream of the stop codon. They were individually inserted into the pJW4303 vector. For pJW4303-tPA-TcdB-C, which had the extra tPA signal peptide sequence upstream of TcdB-C, PCR was conducted from the TcdB-C gene template using the primer F2 (5′ GTCACTTCGCTAGCGTAACAGTAGGAGATGATAAG 3′), incorporating a NheI restriction site, and primer R2 (5′ GAGCTCGGATCCTTATTATTCACTTATTAC 3′), incorporating a BamHI restriction site, and then the tPA-TcdB-C insert was subcloned into pJW4303. All of the DNA vaccine plasmids were produced in large amounts from E. coli HB101 strain with Qiagen Plasmid Mega kit (QIAGEN, 12181) for in vitro transfection and in vivo animal immunization studies.
In vitro expression of target proteins
The expression of toxin A and toxin B DNA vaccines was examined individually by transient transfection of 293 T cells, as previously reported.Citation36 Transfection was done when cells were at approximately 70% confluence on 100 mm dishes by polyethylenimine co-precipitation, using 15 µg of plasmid DNA per dish. At 72 h after transfection, cells were harvested and the supernatants and cell lysates were collected.
Western blot
The 293 T cell transfection samples were subjected to denatured SDS-PAGE and blotted onto PVDF membrane (MILLIPORE, IPVH00010). The membranes were blocked with 5% dried skim milk. Specific animal immune sera raised against individual toxin segments were used as the detecting antibody at 1:500 dilution and incubated at 4°C overnight. The membranes were washed with blocking buffer and reacted with horseradish peroxidase (HRP)-conjugated goat-anti-rabbit IgG (SouthernBiotech, GAR007) at 1:10,000 dilution and incubated at 37°C for 1 h. After the final wash, SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific Inc., 34077) was applied to the membranes for 5 min. Once they were dry, Kodak films were exposed to the membrane and developed in the dark room.
Animal immunizations
NZW rabbits (2 kg body weight) and BALB/c mice (6–8 weeks old) purchased from Shanghai Animal Center, Chinese Academy of Science and Jiangsu Provincial Academy of Agriculture, respectively, were used for immunogenicity studies. They were housed in the Animal Medicine Department of Nanjing Medical University in accordance with approved protocol by the institutional animal use committee.
An electroporator (SCIENTZ-2C) from Scientz Co., Ltd. (SCIENTZ, 07–018) was used for the inoculation of DNA vaccines, as previously described.Citation37 Briefly, following intramuscular injection of plasmid (200 µg per rabbit or 100 µg per mouse divided at two different sites in the quadriceps muscle of the hindlimb), the injection sites were electroporated with the following parameters: 100 V (electric voltage), 60 ms (time constant), and 60 Hz (frequency) for rabbits or 50 V, 30 ms and 30 Hz for mice. A total of four immunizations were given at weeks 0, 2, 4 and 8. Sera were collected prior to and 2 weeks after each immunization.
Enzyme-linked immunosorbent assay (ELISA)
ELISA was conducted to measure the specific antibody responses in sera of immunized rabbits and mice, as previously described.Citation37 The 96-well flat-bottom plates were coated with the supernatant or lysate of 293 T cell transfected with a particular C. difficile toxin segment DNA vaccine plasmid at 1:5 or 1:10 dilution in phosphate buffered saline (PBS), respectively. Samples were incubated at 37°C for 1 h and then washed five times with PBS containing 0.05% Tween-20. Blocking buffer (5% dried skim milk, 0.05% Tween-20 in PBS at pH 7.2) was then added and incubated under the same conditions as above. After another five washes, 100 µl of serially diluted rabbit or mouse sera was added into the duplicate wells and incubated the same as above. After another set of washes, 100 µl biotinylated goat-anti-rabbit IgG or goat-anti-mouse IgG (SouthernBiotech, 4050–08 and 1030–08, respectively) was added, which was diluted to 1:5,000 with PBS containing 4% Whey and 0.5% Tween-20. The plates were incubated the same as above before 100 µl of 1:4000 diluted HRP-conjugated streptavidin (SouthernBiotech, 7100–05) was added after the washes and incubated for another 1 h. After the final wash, the plates were developed with 100 µl 3,3-,5,5-tetramethylbenzidine (TMB) (Sigma, T3405–100TAB) solution per well for 3.5 min. The reaction was stopped by adding 50 µl of 1M H2SO4, and the plates were read at 450 nm. The end titration titer was determined as the highest serum dilution that had an OD value more than twice of that from the negative control serum.
Preparation of bacteria culture filtrate
C. difficile VPI 10463 was inoculated into a BacT/ALERT®SN 259790 culture bottle (bioMérieux, 259790) and incubated at 37°C for 72 h. The culture medium was then centrifuged at 6,000 g for 15 min at 4°C to remove cell debris. The culture medium was then filtered using 0.45 µm filter, and the filtrate was transferred into an Amicon Ultra-15 Centrifugal Filter Unit with Ultracel-100 Membrane (Millipore, UFC810008) and centrifuged at 5000 g for 30 min at 4°C. The final product, concentrated bacteria culture filtrate, was stored at -70°C before use.
Two sandwich ELISA studies were conducted to determine the amount of toxin A and toxin B in concentrated bacteria culture filtrate. A toxin A Detection Kit (Haiyuan Protein Biotech, CD-ToxinA-012a) was used to determine the amount of toxin A. For toxin B, a sandwich ELISA was developed with toxin-specific polyclonal antibodies purchased from Meridian (Meridian Bioscience, Inc., B01246R) for both capturing and detecting purposes. A standard curve was generated using serially diluted commercial toxin A or toxin B products with known concentrations (List Biological Lab, 152C and 155B, respectively). Standard curves were plotted as the standard toxin A or toxin B protein concentration (ng/ml) against the corresponding mean OD value of replicates. A four-parameter curve fit analysis (SOFTmax Pro ver.3) was chosen with a R2 > 0.997. The amount of toxin A or toxin B in concentrated bacteria culture filtrate was determined by interpolating the OD values of test samples into the above standard curves. The amounts of toxin A and toxin B in the concentrated bacterial culture filtrate were 249.76 μg/ml and 212.7 μg/ml, respectively.
In vitro protection study
HT-29 cells were inoculated into a 96-well plate with a concentration of 2 × 104 cells per well and then cultured under 5% CO2 at 37°C for 24 h. The medium was then discarded and the cells were washed with 200 µl pre-heated PBS. Bacteria culture filtrate was serially diluted with Dulbecco's Modified Eagle Medium (DMEM) with 100 U/ml penicillin, 0.1 mg/ml streptomycin and 10% inactivated fetal bovine serum, and then 100 µl of diluted bacteria culture filtrate was added into triplicate wells, while the DMEM medium without any bacteria culture medium concentrate was added into control wells. After 24 h, the cell morphology was observed under a Nikon ECLIPSE TE2000-S microscope (Nikon). The minimal dose of bacteria culture filtrate needed to cause 100% rounding of the cells was defined as 1 cytotoxic unit (CTU). For HT-29 cells, 1 CTU = 1:327,680 dilutions of concentrated bacteria culture filtrate.
For protection studies of antibody against TcdA-C, the HT-29 cells were prepared in the same way as above. Using 1:100 as the initial dilution, 100 µl of serially diluted rabbit sera collected at two weeks after the last immunization and inactivated at 56°C for 30 min were mixed with 4 CTU bacteria culture infiltrate (in 100 µl) and incubated at 37°C for 1 h. The mixture was then added into the triplicate HT-29 cell wells and cultured for another 24 h. Cell morphology was observed under a Nikon ECLIPSE TE2000-S microscope and the neutralization antibody titer was defined as the reciprocal of the highest serum dilution that achieved 100% inhibition of cell rounding.
Similarly, Vero and CHO cells were used to evaluate antibody against rabbit sera elicited by the TcdB-N DNA vaccine with the following difference: the cell density was 1 × 10 4 cells per well for Vero and CHO cell assays. For Vero cells, 1 CTU = 1: 1,310,720 dilutions of concentrated bacteria culture filtrate. For CHO cells, 1 CTU = 1:163,840 dilutions of concentrated bacteria culture filtrate. For protection study in both cell types, 100 µl of 4 CTU concentrated bacteria culture filtrate was used in each well.
In vivo protection study
First, a pre-protection study was conducted in BALB/c mice with serially reduced challenge dose of concentrated bacteria culture filtrate and the absolute lethal dosing (LD100) was determined to be 100 µl of 1:200 diluted concentrated bacteria culture filtrate (containing 125 ng of toxin A and 106 ng of toxin B).
Two types of challenge studies were then conducted. The first study evaluated the protective efficacy of rabbit antibody against TcdA-C using commercial toxin A as the challenging agent. There are three groups: (1) the negative group was injected intraperitoneally (i.p.) with 100ng commercial toxin A (in 100 µl) without any treatment; (2) TcdA-C group was injected i.p. with the mixture of 100 µl of 100 ng commercial toxin A and 11µl rabbit immune sera collected at two weeks after the fourth TcdA-C DNA immunization; and (3) Pre-bleed I group was injected i.p. with the mixture of 100 ng commercial toxin A (in 100 µl) and 11 µl rabbit prebleed sera from the TcdA-C vaccine group. Each group contained 12 mice and the mixture for i.p. injection was pre-incubated at 37°C for 1 h.
In the second type of challenge studies, each BALB/c mouse was injected i.p. with one LD100 (100 µl of 1:200 diluted bacteria culture infiltrate) with or without pre-mixing of rabbit sera as described below. Animals were divided into the following groups: (1) the negative control group (n = 25 mice) did not receive any treatment; (2) Pre-bleed-II group (n = 25 mice) received a mixture of 100 µl diluted bacteria culture filtrate, 5.5 µl pre-immunized rabbit sera from the TcdA-C vaccine group and 5.5 µl pre-immunized rabbit sera from tPA-TcdA-C vaccine group; (3) Pre-bleed-III group (n = 25 mice) received a mixture of 100 µl diluted bacteria culture filtrate, 5.5 µl pre-immunized rabbit sera from the TcdB-N vaccine group and 5.5 µl pre-immunized rabbit serum of TcdA-C vaccine group; (4) Pre-bleed-IV group (n = 20 mice) received a mixture of 100 µl diluted bacteria culture filtrate, 5.5 µl pre-immunized rabbit sera from the TcdB-N vaccine group and 5.5 µl pre-immunized rabbit serum of tPA-TcdA-C vaccine group; (5) the TcdA-C group (n = 25 mice) received a mixture of 100 µl of diluted bacteria culture filtrate with 11 µl rabbit serum collected at two weeks after the fourth TcdA-C immunization; (6) the tPA-TcdA-C group (n = 20 mice) received a mixture of 100 µl of diluted bacteria culture filtrate and 11 µl rabbit serum collected at two weeks after the fourth tPA-TcdA-C immunization; (7) the TcdB-N group (n = 25 mice) received a mixture of 100 µl diluted bacteria culture filtrate and 11 µl rabbit serum collected at two weeks after the fourth TcdB-N immunization; (8) the TcdB-N+TcdA-C group (n = 25 mice) received a mixture of 100 µl diluted bacteria culture filtrate and 11 µl rabbit serum collected at two weeks after the fourth TcdB-N immunization and 11 µl rabbit serum of two weeks after the fourth TcdA-C immunization; (9) the TcdB-N+tPA-TcdA-C group (n = 20 mice) received a mixture of 100 µl diluted bacteria culture filtrate and 11 µl rabbit serum collected at two weeks after the fourth TcdB-N immunization and 11 µl rabbit serum collected at two weeks after the fourth tPA-TcdA-C immunization. All of the mixtures for i.p. injection were pre-incubated at 37°C for 1 h. Survival rates were observed every 12 h for two weeks.
Statistical analysis
Student’s t-test was used to analyze the differences in TcdA- and TcdB-specific binding and neutralizing antibody responses against TcdA or TcdB in immune animal sera between different immunization groups. Fisher’s exact test was used to analyze the passive protection rates in mouse challenge studies. A p value of less than 0.05 was considered significant.
| Abbreviations: | ||
| TMB | = | 3,3-,5,5-tetramethylbenzidine |
| TcdA | = | C. difficile toxin A |
| TcdB | = | C. difficile toxin B |
| CRS | = | cis-acting repressor sequences |
| CTU | = | cytotoxic unit |
| DMEM | = | Dulbecco's Modified Eagle Medium |
| ELISA | = | Enzyme-linked immunosorbent assay |
| HRP | = | horseradish peroxidase |
| INS | = | inhibitory sequences |
| i.p. | = | intraperitoneally |
| NZW | = | New Zealand White |
| PBS | = | phosphate buffered saline |
| RBD | = | receptor binding domain |
| tPA | = | tissue plasminogen activator |
Acknowledgments
This study was supported in part by funding from Jiangsu Province Key Laboratory in Infectious Diseases. Authors would like to thank Dr. Jill Serrano for her critical reading and editing of the manuscript.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Related Research Data
References
- Vonberg RP, Kuijper EJ, Wilcox MH, Barbut F, Tüll P, Gastmeier P, et al, European C difficile-Infection Control Group, European Centre for Disease Prevention and Control (ECDC). Infection control measures to limit the spread of Clostridium difficile. Clin Microbiol Infect 2008; 14:Suppl 5 2 - 20; http://dx.doi.org/10.1111/j.1469-0691.2008.01992.x; PMID: 18412710
- Mutters R, Nonnenmacher C, Susin C, Albrecht U, Kropatsch R, Schumacher S. Quantitative detection of Clostridium difficile in hospital environmental samples by real-time polymerase chain reaction. J Hosp Infect 2009; 71:43 - 8; http://dx.doi.org/10.1016/j.jhin.2008.10.021; PMID: 19041162
- Dubberke ER, Reske KA, Noble-Wang J, Thompson A, Killgore G, Mayfield J, et al. Prevalence of Clostridium difficile environmental contamination and strain variability in multiple health care facilities. Am J Infect Control 2007; 35:315 - 8; http://dx.doi.org/10.1016/j.ajic.2006.12.006; PMID: 17577478
- Jin K, Wang S, Huang Z, Lu S. Clostridium difficile infections in China. Journal of Biomedical Research 2010; 24:411 - 6; http://dx.doi.org/10.1016/S1674-8301(10)60055-3
- Lyerly DM, Saum KE, MacDonald DK, Wilkins TD. Effects of Clostridium difficile toxins given intragastrically to animals. Infect Immun 1985; 47:349 - 52; PMID: 3917975
- Lyras D, O’Connor JR, Howarth PM, Sambol SP, Carter GP, Phumoonna T, et al. Toxin B is essential for virulence of Clostridium difficile. Nature 2009; 458:1176 - 9; http://dx.doi.org/10.1038/nature07822; PMID: 19252482
- Kuehne SA, Cartman ST, Heap JT, Kelly ML, Cockayne A, Minton NP. The role of toxin A and toxin B in Clostridium difficile infection. Nature 2010; 467:711 - 3; http://dx.doi.org/10.1038/nature09397; PMID: 20844489
- Blossom DB, McDonald LC. The challenges posed by reemerging Clostridium difficile infection. Clin Infect Dis 2007; 45:222 - 7; http://dx.doi.org/10.1086/518874; PMID: 17578783
- Hu MY, Katchar K, Kyne L, Maroo S, Tummala S, Dreisbach V, et al. Prospective derivation and validation of a clinical prediction rule for recurrent Clostridium difficile infection. Gastroenterology 2009; 136:1206 - 14; http://dx.doi.org/10.1053/j.gastro.2008.12.038; PMID: 19162027
- Loo VG, Poirier L, Miller MA, Oughton M, Libman MD, Michaud S, et al. A predominantly clonal multi-institutional outbreak of Clostridium difficile-associated diarrhea with high morbidity and mortality. N Engl J Med 2005; 353:2442 - 9; http://dx.doi.org/10.1056/NEJMoa051639; PMID: 16322602
- Maroo S, Lamont JT. Recurrent Clostridium difficile. Gastroenterology 2006; 130:1311 - 6; http://dx.doi.org/10.1053/j.gastro.2006.02.044; PMID: 16618421
- McDonald LC, Killgore GE, Thompson A, Owens RC Jr., Kazakova SV, Sambol SP, et al. An epidemic, toxin gene-variant strain of Clostridium difficile. N Engl J Med 2005; 353:2433 - 41; http://dx.doi.org/10.1056/NEJMoa051590; PMID: 16322603
- Pépin J, Routhier S, Gagnon S, Brazeau I. Management and outcomes of a first recurrence of Clostridium difficile-associated disease in Quebec, Canada. Clin Infect Dis 2006; 42:758 - 64; http://dx.doi.org/10.1086/501126; PMID: 16477549
- Surawicz CM. Reining in recurrent Clostridium difficile infection--who’s at risk?. Gastroenterology 2009; 136:1152 - 4; http://dx.doi.org/10.1053/j.gastro.2009.02.023; PMID: 19250653
- Giannasca PJ, Warny M. Active and passive immunization against Clostridium difficile diarrhea and colitis. Vaccine 2004; 22:848 - 56; http://dx.doi.org/10.1016/j.vaccine.2003.11.030; PMID: 15040937
- Belyi IF, Varfolomeeva NA. Construction of a fusion protein carrying antigenic determinants of enteric clostridial toxins. FEMS Microbiol Lett 2003; 225:325 - 9; http://dx.doi.org/10.1016/S0378-1097(03)00560-3; PMID: 12951260
- Pavliakova D, Moncrief JS, Lyerly DM, Schiffman G, Bryla DA, Robbins JB, et al. Clostridium difficile recombinant toxin A repeating units as a carrier protein for conjugate vaccines: studies of pneumococcal type 14, Escherichia coli K1, and Shigella flexneri type 2a polysaccharides in mice. Infect Immun 2000; 68:2161 - 6; http://dx.doi.org/10.1128/IAI.68.4.2161-2166.2000; PMID: 10722615
- Sauerborn M, Leukel P, von Eichel-Streiber C. The C-terminal ligand-binding domain of Clostridium difficile toxin A (TcdA) abrogates TcdA-specific binding to cells and prevents mouse lethality. FEMS Microbiol Lett 1997; 155:45 - 54; http://dx.doi.org/10.1111/j.1574-6968.1997.tb12684.x; PMID: 9345763
- Lyerly DM, Johnson JL, Frey SM, Wilkins TD. Vaccination against lethalClostridium difficile enterocolitis with a nontoxic recombinant peptide of toxin A. Curr Microbiol 1990; 21:29 - 32; http://dx.doi.org/10.1007/BF02090096
- Wren BW, Russell RR, Tabaqchali S. Antigenic cross-reactivity and functional inhibition by antibodies to Clostridium difficile toxin A, Streptococcus mutans glucan-binding protein, and a synthetic peptide. Infect Immun 1991; 59:3151 - 5; PMID: 1715320
- Ward SJ, Douce G, Figueiredo D, Dougan G, Wren BW. Immunogenicity of a Salmonella typhimurium aroA aroD vaccine expressing a nontoxic domain of Clostridium difficile toxin A. Infect Immun 1999; 67:2145 - 52; PMID: 10225867
- Ward SJ, Douce G, Dougan G, Wren BW. Delivery of non-toxic fragments of Clostridium difficile toxin A to the mucosal immune system. Reviews in Medical Microbiology 1997; 8:S37; http://dx.doi.org/10.1097/00013542-199712001-00016
- Ryan ET, Butterton JR, Smith RN, Carroll PA, Crean TI, Calderwood SB. Protective immunity against Clostridium difficile toxin A induced by oral immunization with a live, attenuated Vibrio cholerae vector strain. Infect Immun 1997; 65:2941 - 9; PMID: 9199470
- Gardiner DF, Rosenberg T, Zaharatos J, Franco D, Ho DD. A DNA vaccine targeting the receptor-binding domain of Clostridium difficile toxin A. Vaccine 2009; 27:3598 - 604; http://dx.doi.org/10.1016/j.vaccine.2009.03.058; PMID: 19464540
- Seregin SS, Aldhamen YA, Rastall DP, Godbehere S, Amalfitano A. Adenovirus-based vaccination against Clostridium difficile toxin A allows for rapid humoral immunity and complete protection from toxin A lethal challenge in mice. Vaccine 2012; 30:1492 - 501; http://dx.doi.org/10.1016/j.vaccine.2011.12.064; PMID: 22200503
- Drudy D, Fanning S, Kyne L. Toxin A-negative, toxin B-positive Clostridium difficile. Int J Infect Dis 2007; 11:5 - 10; http://dx.doi.org/10.1016/j.ijid.2006.04.003; PMID: 16857405
- Drudy D, Harnedy N, Fanning S, Hannan M, Kyne L. Emergence and control of fluoroquinolone-resistant, toxin A-negative, toxin B-positive Clostridium difficile. Infect Control Hosp Epidemiol 2007; 28:932 - 40; http://dx.doi.org/10.1086/519181; PMID: 17620240
- Xu G, Wang S, Zhuang L, Hackett A, Gu L, Zhang L, et al. Intramuscular delivery of a cholera DNA vaccine primes both systemic and mucosal protective antibody responses against cholera. Vaccine 2009; 27:3821 - 30; http://dx.doi.org/10.1016/j.vaccine.2009.04.008; PMID: 19443090
- Albesa-Jové D, Bertrand T, Carpenter EP, Swain GV, Lim J, Zhang J, et al. Four distinct structural domains in Clostridium difficile toxin B visualized using SAXS. J Mol Biol 2010; 396:1260 - 70; http://dx.doi.org/10.1016/j.jmb.2010.01.012; PMID: 20070948
- Lowy I, Molrine DC, Leav BA, Blair BM, Baxter R, Gerding DN, et al. Treatment with monoclonal antibodies against Clostridium difficile toxins. N Engl J Med 2010; 362:197 - 205; http://dx.doi.org/10.1056/NEJMoa0907635; PMID: 20089970
- Babcock GJ, Broering TJ, Hernandez HJ, Mandell RB, Donahue K, Boatright N, et al. Human monoclonal antibodies directed against toxins A and B prevent Clostridium difficile-induced mortality in hamsters. Infect Immun 2006; 74:6339 - 47; http://dx.doi.org/10.1128/IAI.00982-06; PMID: 16966409
- Ibsen PH. The effect of formaldehyde, hydrogen peroxide and genetic detoxification of pertussis toxin on epitope recognition by murine monoclonal antibodies. Vaccine 1996; 14:359 - 68; http://dx.doi.org/10.1016/0264-410X(95)00230-X; PMID: 8735545
- Lu S. Heterologous prime-boost vaccination. Curr Opin Immunol 2009; 21:346 - 51; http://dx.doi.org/10.1016/j.coi.2009.05.016; PMID: 19500964
- Wang S, Kennedy JS, West K, Montefiori DC, Coley S, Lawrence J, et al. Cross-subtype antibody and cellular immune responses induced by a polyvalent DNA prime-protein boost HIV-1 vaccine in healthy human volunteers. Vaccine 2008; 26:1098 - 110; http://dx.doi.org/10.1016/j.vaccine.2007.12.024; PMID: 18243434
- Ledgerwood JE, Wei CJ, Hu Z, Gordon IJ, Enama ME, Hendel CS, et al, VRC 306 Study Team. DNA priming and influenza vaccine immunogenicity: two phase 1 open label randomised clinical trials. Lancet Infect Dis 2011; 11:916 - 24; http://dx.doi.org/10.1016/S1473-3099(11)70240-7; PMID: 21975270
- Pear WS, Nolan GP, Scott ML, Baltimore D. Production of high-titer helper-free retroviruses by transient transfection. Proc Natl Acad Sci USA 1993; 90:8392 - 6; http://dx.doi.org/10.1073/pnas.90.18.8392; PMID: 7690960
- Wang S, Zhang C, Zhang L, Li J, Huang Z, Lu S. The relative immunogenicity of DNA vaccines delivered by the intramuscular needle injection, electroporation and gene gun methods. Vaccine 2008; 26:2100 - 10; http://dx.doi.org/10.1016/j.vaccine.2008.02.033; PMID: 18378365