Abstract
A significant hurdle in vaccine development for many infectious pathogens is the ability to generate appropriate immune responses at the portal of entry, namely mucosal sites. The development of vaccine approaches resulting in secretory IgA and mucosal cellular immune responses against target pathogens is of great interest and in general, requires live viral infection at mucosal sites. Using HIV-1 and influenza A antigens as models, we report here that a novel systemically administered DNA vaccination strategy utilizing co-delivery of the specific chemokine molecular adjuvant CCL25 (TECK) can produce antigen-specific immune responses at distal sites including the lung and mesenteric lymph nodes in mice. The targeted vaccines induced infiltration of cognate chemokine receptor, CCR9+/CD11c+ immune cells to the site of immunization. Furthermore, data shows enhanced IFN-λ secretion by antigen-specific CD3+/CD8+ and CD3+/CD4+ T cells, as well as elevated HIV-1-specific IgG and IgA responses in secondary lymphoid organs, peripheral blood, and importantly, at mucosal sites. These studies have significance for the development of vaccines and therapeutic strategies requiring mucosal immune responses and represent the first report of the use of plasmid co-delivery of CCL25 as part of the DNA vaccine strategy to boost systemic and mucosal immune responses following intramuscular injection.
Introduction
Plasma DNA has a number of advantages that make it an attractive vaccine platform.Citation1 First, it is a non-live, non-replicating, non-spreading and non-viral platform that to date has had an excellent safety profile. Neither pre-existing immunity nor the development of neutralizing antibodies against the plasmid backbone is an issue for DNA vaccines, which allow for boosting of specific immune responses after multiple immunizations. Plasmid DNA is also simple to manipulate and produce compared with live vector-based vaccines, including those of viral origin. Furthermore, it is a stable platform that may not require a cold-chain for storage. DNA vaccines have been shown to elicit humoral and cellular responses and confer protection in small animal models and in larger species to some degree.Citation1-Citation5
One of the limitations of DNA vaccines is that they are primarily a systemic immunization strategy. Reports have suggested that the route of vaccination determines the ability to generate a mucosal immune response, with elicited T and B cells preferentially homing to sites where they were induced. Accordingly, several investigators have utilized DNA immunization in conjunction with a mucosal vaccination route, allowing for the generation of immune responses at both mucosal and systemic sites.Citation6,Citation7 The inclusion of genes encoding cytokines or chemokines in a vaccination strategy can alter the magnitude, duration and nature of the immune response, and these entities have been tested as vaccine adjuvants.Citation8-Citation13 Mucosal-targeting adhesion molecules have been used to direct antigenic protein immune responses to the gut.Citation14-Citation18
We and others have reported redirection of systemic immune cells in vivo by DNA vaccination using chemokines as part of a DNA-encoded vaccine cocktail.Citation14,Citation15,Citation19-Citation21 Recent work further confirmed and elegantly extended these findingsCitation22,Citation23 through modification of vaccine-induced immune cell trafficking by utilizing chemokines (immune trafficking signals) to attract peripheral immune cell populations. The further modulation of immune responses through the chemokine network appears interesting.
The identification of chemokines/chemokine receptors expressed within mucosal and epithelial tissue has enhanced our understanding of migration and localization of tissue-specific antibody secreting cell (ASCs), as well as naïve, effector and memory T lymphocyte recirculation to mucosal and cutaneous sites. Mucosal homing of memory B and T lymphocytes is mediated by a unique combination of adhesion molecules, the mucosal chemokines, CCL25 (TECK), CCL27 (CTACK) and mucosal-associated epithelial chemokine, CCL28 (MEC), as well as the chemokine receptors CCR9 and CCR10,Citation24-Citation27 supporting the importance of these molecules in mucosal immunity. It has been demonstrated that CCR9 expression is upregulated on activated T cells and is essential for trafficking of CCR9+ T cells in the lamina propria and intraepithelium of small intestine.Citation28 At this site, primed and activated mucosal T cells reside ready for antigen re-challenge.Citation29-Citation31 In addition, CCL25 functions synergistically with CCL28 (MEC) to recruit IgA+ plasmablasts expressing CCR9 and CCR10 to small intestinal lamina propria by enhancing their 〈4 integrin-dependent adhesion to mucosal addressin cell adhesion molecule-1 and vascular cell adhesion molecule-1 (VCAM-1), supporting the importance of these molecules in mucosal immunity.Citation24,Citation32 In addition to T cells, an essential role for CCR9 in the homing of plasmacytoid dentritic cells (pDCs) to the intestine under homeostatic and inflammatory conditions has been reported and supports an important role for intestinal pDCs for the rapid mobilization of lamina propria DCs.Citation33 Chemokines and their corresponding receptors are main players regulating the homing of immune-effector cells from inductive sites to mucosal effector sites. In this respect, CCL25 could represent a suitable candidate as a mucosal molecular adjuvant.
We report here that co-delivery of plasmid expressing CCL25 with an antigenic construct for HIV-1gag or influenza A/PR/8/34 hemagglutinin resulted in the induction of elevated levels of antigen-specific inflammatory cytokines in the periphery as well as mucosal sites including both the lung and gut-associated lymphoid tissue (GALT). Furthermore, examination of antigen-specific IgA production at mucosal sites or in feces was increased by this unique DNA plasmid adjuvant. In an influenza challenge model system, CCL25 co-immunization resulted in the induction of long-lived IgA, neutralizing antibodies and protection of mice from morbidity and mortality associated with a lethal mucosal viral challenge. Taken together, our results suggest that the use of the CCR9 ligand, CCL25, as a DNA vaccine adjuvant to influence vaccine responses at mucosal sites appears physiologically relevant, and should be tested in a larger animal model of mucosal pathogen challenge.
Results
Construction and expression of mucosal chemokine plasmid.
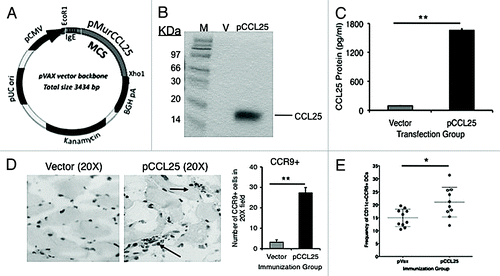
We had observed that CCR9 was expressed on lymphocytes from the spleen, Peyer’s patches and mesenteric lymph node of wild type Balb/c but not CCR9 knockout miceCitation34 (data not shown), suggesting that this receptor could be targeted during an antigen-specific immune response. Furthermore, we hypothesized that the CCR9 ligand may modulate immune responses in a unique fashion. Therefore, we constructed a plasmid form of pCCL25 and tested its expression prior to its use as an adjuvant in a vaccination regimen. The murine form of CCL25 (435 base pairs) was generated by PCR using cDNA synthesized from RNA purified from murine thymus. Chemokine cDNA was inserted into the multiple cloning site of the pVAX1 vector (). To test the expression of the protein encoded by the chemokine construct, we analyzed the protein expression through an in vitro translation assay using specific anti-mouse CCL25 antibody (). In order to confirm expression in vitro, in separate experiments, we transfected the RD cell line with the vector control or the chemokine plasmid, and collected supernatants 48 h after transfection. We measured levels of secreted CCL25 protein by the RD cell lines using ELISA and detected 16,000 pg/ml of CCL25 (). In addition, CCR9+ lymphocytes collected from mucosal tissues [lung, mesenteric lymph nodes (MLN) or Peyer’s patch/colonic cryopatch] migrated in response to supernatants from chemokine-transfected cells in a chemotaxis transwell experiment (data not shown). Taken together, these data demonstrate that the plasmid expression vector expresses the specific and functional CCL25 chemokine protein.
Figure 1. Expression of bioactive chemokines from plasmid encoded mucosal chemokine and the induction of infiltration, after intramuscular injection, by cognate receptor positive cells (A). All genes were cloned into the multiple cloning region of the expression vector pVAX (kanamycin resistance) using the restriction enzyme sites, EcoR1 and Xho1. (B) Expression of pCCL25 was confirmed by a T7 coupled transcription/translation reticulocyte lysate system. The blotting gel shows size markers (designated M, lane 1), pVAX background control protein (V, lane 2), 14.2 kDa CCL25 protein (lane 3). (C) Expression of bioactive chemokine protein translated from the plasmid forms of CCL25. ELISA was performed using supernatants from pCCL25-transfected RD cells (pg/ml). Vector background control is included (gray bar) vs. pCCL25 (black bar). Data are shown as pg/ml of chemokine protein ± SD of triplicate wells. (D) Infiltration of CCR9 positive cells induced following intramuscular injection of 100 μg of pCCL25. Immunohistochemical staining of quadriceps sections 7 d post immunization with 100 μg of vector backbone control (left panel) or pCCL25. Infiltrating cells were enumerated following a visual count by microscope of brown positive cells over 6 (20×) fields, and the total number of CCR9+ cells was averaged and shown as ± SD for vector (gray bar) and pCCL25 injection (black bar). (E) Frequency of CD11c+ cells that express CCR9 in the popliteal and inguinal DLNs of individual immunized mice in vector backbone immunized (solid black symbols) vs. pCCL25 immunized mice (open circles). (*p < 0.05, **p < 0.01, for comparisons between vector backbone pVAX or pCCL25 immunized groups.) Data are representative of three independent experiments, and in panel (E), ± SEM is shown because individual mouse DLNs were tested.
In vivo bioactivity of chemokine protein is generated from plasmid CCL25.
We next initiated studies to determine whether intramuscular delivery of pCCL25 could result in increased recruitment of the cognate receptor (CCR9) positive cells to the site of vaccine inoculation. Groups of Balb/c mice (n = 4 per group) received intramuscular injections into the quadriceps of 100 μg of either a vector backbone plasmid or pCCL25 constructs. The injected muscles were excised on day 7 following immunization, and 5 μm serial sections were examined quantitatively for the presence of local cellular inflammatory infiltrates. The muscle was sectioned and immunohistochemistry was performed to visualize infiltrating cell expression of the chemokine receptor CCR9 (), followed subsequently by quantitation of CCR9+ cells (). Values were determined by visual count of 6 independent 20× fields from the experimental slides viewed randomly under a microscope and positive cells (brown DAB substrate positive) were enumerated, averaged and graphed as the total number of CCR9+ cells per 20× field. Few CCR9 positive cells were observed following injection of the vector backbone alone, while immunization with pCCL25 resulted in larger inflammatory cell clusters that expressed CCR9. Quantitation of these inflammatory infiltrates demonstrated that intramuscular administration of pCCL25 resulted in a greater than 7-fold increase in the recruitment of CCR9+ inflammatory cells to the muscle compared with vector backbone co-delivery alone ()(i.e., 25.5 ± 3.2-fold vs. 3.2 ± 1.2-fold). In addition, as shown in , when we examined the frequency (15.0% ± 1.06 SEM vector backbone control vs. 21.0% ± 1.82 SEM pCCL25, p = 0.01) and absolute numbers (3642 ± 502 SEM vector backbone control vs. 6,706 ± 1,067 SEM pCCL25, p = 0.018, data not shown) of CD11c+ cells that express CCR9 in the popliteal and inguinal DLNs of pCCL25-immunized mice, we observe a significant elevation (n = 10 independent DLN specimens). Data shown represent frequencies of CD11c+/CCR9+ cells (± SEM) present in the DLN of individual vaccinated mice.
Systemic co-delivery of pCCL25 enhances antigen-specific cytokine secretion by T lymphocytes from the spleen or gut.
We next sought to examine whether systemic delivery of mucosal chemokines co-delivered with pHIV-1gag results in enhanced antigen-specific cellular immunity by antigen-specific T lymphocytes. We analyzed antigen-specific cytokine secretion by CD4 and CD8 T cells in the secondary lymphoid organs (using depletion beads) by ELISpot assay, and by lymphocytes isolated by the MLN using intracellular flow analysis. Female Balb/c mice, aged 6–8 weeks, were co-immunized intramuscularly 2 weeks apart for a total of three injections with vector control, pHIV-1gag or pHIV-1gag plus 100 μg of plasmid CCL25. In response to stimulation with HIV-1gag consensus B peptide pools (pools 1 to 4), the optimal doses of chemokine plasmids were determined to be 100 μg per injection (data not shown), and these amounts resulted in augmented levels of HIV-1-specific IFN-λ secretion by T lymphocytes. As shown in , co-immunization with the CCR9 ligand, pCCL25, resulted in increased levels of HIV-1gag-specific IFN-λ as measured by ELISpot. The pCCL25 group exhibited 847 (± 58.2 SD) IFN-y spot forming cells (SFCs) per million splenocytes compared with 333 SFCs (± 76 SD) for pHIV-1gag immunized only (). Pool 2 contains the described dominant CD8+ T cell epitope peptide (for Balb/c mice, amino acids 197 to 211, AMQ MLK ETI NEE AAE) and the subdominant peptide is in pool 3 (gag amino acids 293–307, VDR FYK TLR AEQ ASQ).Citation3,Citation35 Following CD8+ T cell depletion, enhanced IFN-λ secretion was mostly abrogated (847 SFCs to 132 SFCs), suggesting CD8+ T cells secreted the majority of the IFN-λ observed in the ELISpot assay ().
Figure 2. Induction of mucosal antigen-specific IFN-λ in secondary lymphoid organs and MLNs after mucosal chemokine systemic co-immunization. (A) Mice (n = 4 per group) were immunized intramuscularly three times, with each injection separated by 2 weeks. Splenocytes were harvested from vector control, pHIV-1gag, pHIV-1gag/pCCL25, 1 week following the last injection. Splenocytes from vaccinated animals from each experimental group were pooled and were cultured overnight in the presence of medium (gray bar, negative control) or 10 μg/ml HIV-1 peptide pools 1 through 4 (hatched bars). The solid black bar represents the total number of HIV-1-specific IFN-λ SFCs per million splenocytes for all 4 HIV-gag pools. (B) CD8+ T cell depletion partially abrogates HIV-1gag-specific IFN-λ secreted by the lymphocytes from immunized mice. Total HIV-1gag-specific IFN-λ SFCs are shown as black bars and following CD8+ T cell depletion (gray bars). Values represent SFCs per million splenocytes and error represents the mean ± SD of the triplicate cultures and are representative of 6 independent experiments. (C) pCCL25 co-immunization enhances HIV-1-specific IFN-λ secretion by CD3+/CD4 and CD3+/CD8 MLN T lymphocytes. Mice were immunized intramuscularly three times, with each injection separated by 2 weeks. MLNs were harvested from vector control, pHIV-1gag, pHIV-1gag/pCCL25, 1 week following the last injection. Mucosal lymphocytes were harvested and stimulated with either medium background control, HIV-1gag peptides or PMA and ionomycin. Cells were stained as described in “Methods” and 50,000 CD3+ live lymphocytes (as determined by the LIVE/DEAD stain) were collected. Total IFN-λ secreting CD3+/CD4 (hatched upper stack) or CD3+/CD8 (gray lower stack) are graphed as the mean ± SEM of individual mice for vaccination groups. Responses from the negative control wells were subtracted from the antigenic stimulations prior to graphing (*p ≤ 0.05, ** p ≤ 0.01, for statistical comparisons between pCCL25 co-immunized groups vs. pHIV-1gag alone for similar treatment conditions). Data are representative of three independent experiments.
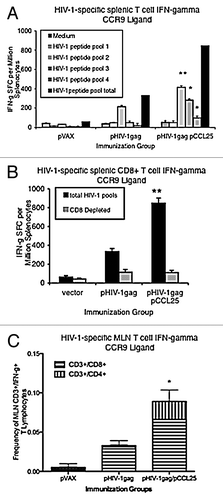
Studies were subsequently performed to test whether co-immunization with the CCL25 chemokine plasmid could modulate the induction of antigen-specific cytokine secretion at mucosal sites. Following vaccination, MLNs were harvested and lymphocytes were stimulated with either medium (for background control) or the HIV-1gag peptide pools. Intracellular flow analysis was performed to measure the frequency of antigen-specific CD3+/CD4+/CD8+ lymphocytes from vaccinated mice that secrete IFN-λ in response to HIV-1gag restimulation (). Values represent the percent of CD3+ T cell (CD4+ and CD8+) lymphocytes that secrete IFN-λ after background medium stimulation was subtracted and the error represents ± SEM of individual mice within each experimental group. While the magnitude of the responses is low, we were able to detect augmented HIV-1gag-specific IFN-λ (0.1%) by CD8+ T and CD4+ T lymphocytes located in the MLNs in response to pCCL25 co-immunization ().
pCCL25 co-immunization enhances HIV-1gag-specific sera and fecal IgA.
We next tested whether co-immunization with the mucosal chemokine, pCCL25, enhanced IgA secretion by ELISA () and B-cell ELISpot (). Mice were co-immunized intramuscularly with vector control, pHIV-1gag or pHIV-1gag in combination with pCCL25 as described in the “Materials and Methods.” The three immunizations were performed at 2-week intervals, and samples were harvested 10 d after the last boost. indicate the average fold increase in the anti-HIV-1gag IgA responses when the chemokine adjuvanted group is compared with antigenic plasmid immunization alone. A modest but statistically significant increase (1.5-fold) of IgA was observed in the peripheral blood (sera) of pCCL25 immunized mice (). In fecal extracts, antigenic plasmid immunization resulted in a low level of HIV-1gag-specific immunoglobulin. However, when the antigenic plasmid was combined with pCCL25, co-immunization induced a 1.8-fold increase in HIV-1gag-specific fecal IgA (290.21 μg/ml) ().
Figure 3. Augmented secretory IgA in the periphery and in the GALT following systemic co-immunization with mucosal chemokine. (A–B) The average fold increase of anti-HIVgag-specific IgA elicited by the chemokine immune adjuvants over pHIV-1gag antigenic plasmid alone is shown in panel A (fecal IgA) and panel B (sera IgA). Data generated from 6 independent experiments of 4 mice per group were averaged and fold increases were determined. Levels of HIV-1 specific IgA in fecal extract (A) or sera (B) harvested 10 d following the last immunization were determined by an ELISA assay against HIV-1gag p24 protein. Quantitation of fecal and serum IgA (μg/ml) was determined using a recombinant IgA standard of known concentration as the standard curve, and fold increase values were determined. (C–D) Mucosal chemokine enhanced IgA antibody secretion cell frequency in Peyer’s patches (C) and in the spleen (D). Whole Peyer’s patch (C) or splenocyte (D) immune cells purified from vector, pHIV-1gag or pCCL25 co-immunized mice were placed in an HIV-1gag p24 protein coated, 96 well ELISpot blocked plate and incubated for 5 h. Secreted anti-HIV-1gag IgA by antibody secreting plasmablasts was captured on plate bound P24, counted using an ELISpot reader and graphed as HIV-1-specific IgA ASCs per million Peyer’s patch B cells or splenocytes. Error represents the mean ± SD of triplicate wells (*p ≤ 0.05, ** p ≤ 0.01, for statistical comparisons between pCCL25 co-immunized groups vs. pHIV-1gag alone for similar treatment conditions). Data are representative of three independent experiments.
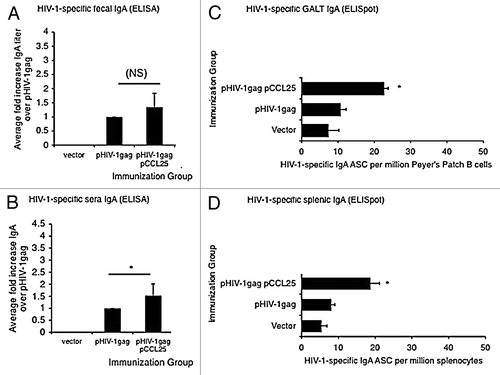
We confirmed the presence of augmented secretion of HIV-1gag-specific IgA by B lymphocytes following systemic delivery of pCCL25 in the GALT or spleen by B cell ELISPOT (, respectively) from pHIV-1gag co-immunized Balb/c mice. As indicated in , pCCL25 co-immunization resulted in 23 ± 1.154 SD ASCs (p = 0.016) in cells isolated from mouse intestine compared with the levels measured in the antigen-only (pHIV-1gag) immunized group (11 ± 1.5 SD ASC). When the frequency of IgA secreted by splenic B cells was measured (), we observed a 2-fold increase in IgA when animals were co-immunized with pCCL25 (19 ± 2.51 SD ASCs, p = 0.057) compared with pHIV-1gag alone (8 ± 1.0 SD ASCs) (). Taken together, these data support a role for the CCR9 ligand, CCL25, as a modest inducer of antigen-specific IgA secretion in a mouse model. Of note, we did not detect any increases in total IgG, IgA or IgM antibodies in the CCL25 adjuvanted groups (data not shown), supporting the specificity of these effects on antigenic responses.
Co-delivery of pCCL25 enhances influenza APR/8/34 hemagglutinin-specific cytokine secretion by T lymphocytes in the lung.
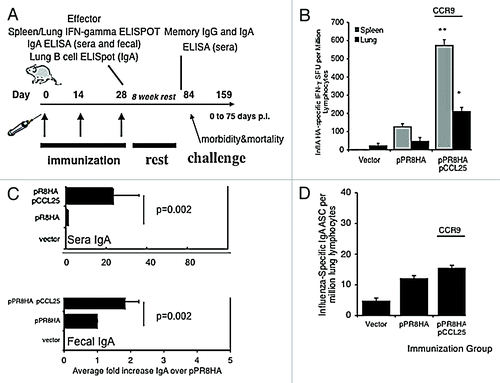
We next examined the ability of intramuscular delivery of the CCR9 chemokine ligand plasmid to modulate antigen-specific cytokine secretion by T cells in an antigen system that could also be evaluated in challenge model studies. We were interested in influenza A/PR/8/34 hemagglutinin since vaccine-induced hemagglutinin-specific T cell and antibody responses have an impact on influenza A infection in the lung. The role that CCR9 ligands play during influenza infection is unknown. As shown in , Balb/c female mice, aged 6–8 weeks, were co-immunized three times intramuscularly with 133 μg of control vector, 33 μg of the hemagglutinin gene from influenza A/PR/8/34 (pPR8HA) plus 100 μg of pVAX backbone control DNA, or 33 μg of the pPR8HA in combination with 100 μg of pCCL25. Each injection was separated by a 2-week interval. Co-immunization with pCCL25 significantly enhanced HA-specific IFN-λ in the spleen when compared with antigenic plasmid alone (). Interestingly, co-immunization with 100 μg of pCCL25 (210 ± 23 SD SFC) significantly enhanced (p < 0.01) the number of HA-specific lung T lymphocytes that secrete IFN-λ compared with pPR8HA immunization alone (47 ± 9.0 SD SFC) ().
Figure 4. pCCL25 immune adjuvant-induced CTL and IgA immunity specific against influenza A/PR/8/34 in a mouse model of mucosal lung infection. (A) Schedule for influenza A (PR8) immunization, immune analysis and lethal mucosal challenge. Mice (n = 10 per group) were immunized as previously described with 33 μg of HA or in combination with our pCCL25. Effector immune responses were measured 1 week following the third immunization, and long-lived responses were examined 8 weeks after the final boost (pre-challenge). (B) Mucosal chemokines enhanced the secretion of IFN-λ influenza specific A/PR/8/34 by lung and splenic T cells. Lung lymphocytes (black bar) or splenocytes (gray bar) were harvested from immunized mice and utilized in an IFN-λ ELISpot assay stimulated with either medium control or the CD8+ T cell epitope peptide encoding influenza A/PR/8/34 (H1N1) hemagglutinin (IYSTVASSL amino acid 518–526). Values depicted in panel (B) are resulting values after medium is subtracted and error represents the mean ± SD of triplicate wells (*p < 0.05, **p < 0.01 for statistical comparisons of chemokine/pPR8HA groups with pPR8HA antigenic vector alone). (C) pCCL25 co-immunization augments mucosal-specific IgA in sera and fecal extracts. Pooled sera and fecal pellets from 4 mice per group were analyzed, by ELISA, for the presence of influenza A/PR/8/34 hemagglutinin specific IgA and data generated from six independent experiments were averaged and shown in panel (C). The fold increase observed from vaccinated sera (upper panel) and feces (lower panel) IgA by ELISA using a recombinant IgA standard of known concentration and the fold increase in IgA levels are shown, and statistical analysis was performed between the chemokine adjuvanted groups in relation to pPR8HA antigenic plasmid alone across 6 independent experiments. (D) pCCL25 co-immunization does not increase the frequency of IgA secreting plasmablasts in the lung. Lung lymphocytes isolated from vector, pPR8HA or pCCL25 co-immunized (perfused) mice were added to the wells of UV-inactivated influenza A/PR/8/34-coated, 96 well ELISpot blocked plates and incubated for 5 h. Secreted anti-HA IgA plasmablasts were captured on plate bound influenza A, counted using an ELISpot reader and graphed as influenza-specific IgA ASCs per million lung lymphocytes. Error represents the mean ± SD between triplicate wells. (The data are a representation of three independent experiments.)
Co-immunization with pCCL25 and pPR8HA induces humoral immunity specific against influenza A/PR/8/34 hemagglutinin.
One week following the last injection, sera or fecal extracts were collected and processed for ELISA in order to measure influenza A-specific immunoglobulin ( IgA data). Immunization with antigenic plasmid (50 μg pPR8HA) alone resulted in modest levels of influenza A-specific IgG in the sera (96.15 μg/ml ± 13.4 SD) or fecal extracts (402 μg/ml ± 29.7 SD). However, serum IgG levels analyzed following co-immunization with pCCL25 resulted in (155.1 μg/ml ± 13.9 SD), a 1.6-fold increase over antigen only (data for IgG not shown). For IgA, co-immunization with antigenic plasmid (50 μg pPR8HA) alone resulted in negligible levels of influenza A-specific IgA in the sera (0.37 μg/ml ± 0.08 SD) or fecal extract (16.1 μg/ml ± 0.33 SD). However, serum IgA levels analyzed following co-immunization with pCCL25 resulted in (7.89 μg/ml ± 1.07 SD), a 21-fold increase over antigen only and a 2-fold increase in IgA levels against influenza A in feces (31.6 μg/ml ± 2.1 SD) (). The modest increase in mucosal IgA observed following pCCL25 co-immunization as measured by ELISA (feces) was similar to findings that pCCL25 co-immunization did not enhance lung antibody secreting cell production of anti-influenza IgA as measured by ELISpot assay ().
Influenza challenge studies.
To test whether the chemokine-induced enhancement of influenza A-specific lung lymphocyte cytokine and IgA secretion reaches protective physiological levels, we performed influenza A/PR/34/8 lethal intranasal challenge. Our previous studies suggested that 10 LD50 influenza A/PR/8/34 resulted in 100% lethality in naïve (i.e., unvaccinated) Balb/c mice. Conversely, greater than 80% and less than 10% lethality occurred in mice receiving 33 μg or 50 μg of the pPR8HA vaccine, respectively.Citation3 Prior to challenge and following an 8-week rest after immunization with 33 μg of pPR8HA and 100 μg of pCCL25, serum was harvested from immunized mice and an ELISA was performed, showing that the adjuvant boosted long-lived influenza A-specific IgG and IgA levels () of individual mice. Using individual serum samples from the rested mice prior to challenge (n = 10), we performed a neutralization assay (6 replicate wells each) (). The serum dilution at which 2000TCID50/ml of influenza A/PR/8/34 is neutralized is depicted in for each study group. A representative well showing CRBC (chicken red blood cell) agglutination (influenza A/PR/8/34, “fuzzy” well) for vector and antigenic plasmid immunization alone as well as representative wells showing CRBC pooling (well contains influenza A/PR/8/34 neutralized virus) for pCCL25 are shown in . The sera dilution at which 2000TCID50/ml of influenza A/PR/8/34 can be neutralized by pPR8HA plasmid alone is 1 to 36, while pCCL25 is 1 to 144, supporting the significantly enhanced ability of the CCR9 ligand to enhance humoral immunity when co-immunized as a plasmid adjuvant with influenza antigenic constructs.
Figure 5. pCCL25 co-immunization induces long-lived IgG, IgA, neutralizing antibody and protects mice against influenza A/PR/8/34 lethal mucosal challenge. (A) Influenza specific long-lived IgG and IgA titers were determined prior to challenge. Individual sera samples were harvested from mice from each immunization group (n = 10 mice per group) and (ng/ml) levels of pPR8HA-specific IgG (gray bar) and IgA (black bar) were determined by ELISA using a recombinant standard of known concentration. Error represents ± SEM since individual mouse sera samples were analyzed (*p < 0.05 for statistical comparisons between chemokine plasmid adjuvanted groups, n = 10 mice per experimental group, vs. n = 10 pPR8HA alone). (B) Co-immunization with pCCL25 elicits anti-influenza A/PR/8/34 neutralizing antibodies. Serial 4-fold dilutions of individual serum samples from immunized mouse groups are incubated with influenza A/PR/8/34 (100 TCID50 virus per well), and results from the chicken erythrocyte sedimentation assay are shown as the serum dilution at which 2000TCID50/ml of influenza A/PR/8/34 can be neutralized. Data represent individual mouse sera (n = 10) in 6 replicates per dilution for each immunized mouse group. Representative wells of non-neutralized virus (agglutination) are shown for control and pR8HA immunized groups, and a single well depicting neutralized virus (no agglutination or a pool of red blood cells at bottom of well) representing pCCL25 co-immunized group. (C–D) Systemic co-immunization with pCCL25 protects mice from influenza A/PR/8/34 intranasal challenge. Average percent weight loss (morbidity) and death (survival) in groups of mice immunized and challenged were as depicted in the immunization schedule shown in . Vector immunized mice (diamonds) lost weight rapidly (C) and 100% (n = 10) died by day 9 post infection (D). Mice that were immunized with pPR8HA (triangles) alone lost an average 18% body weight (C) by day 10 and 80% died (D) by day 10 post challenge. The two surviving mice in the pPR8HA (triangles) immunized group regained weight and survived. Mice immunized with the CCR9 ligand, pCCL25, had a slight reduction in weight (4.7% weight loss) (squares, (C), but all ultimately survived challenge [squares, in panel (D)]. Using log rank analysis, pVAX vs. pR8HA (p = 0.0005), pVAX vs. CCL25 (p < 0.0001), pR8HA vs. pR8HA/CCL25 (p = 0.0003).
![Figure 5. pCCL25 co-immunization induces long-lived IgG, IgA, neutralizing antibody and protects mice against influenza A/PR/8/34 lethal mucosal challenge. (A) Influenza specific long-lived IgG and IgA titers were determined prior to challenge. Individual sera samples were harvested from mice from each immunization group (n = 10 mice per group) and (ng/ml) levels of pPR8HA-specific IgG (gray bar) and IgA (black bar) were determined by ELISA using a recombinant standard of known concentration. Error represents ± SEM since individual mouse sera samples were analyzed (*p < 0.05 for statistical comparisons between chemokine plasmid adjuvanted groups, n = 10 mice per experimental group, vs. n = 10 pPR8HA alone). (B) Co-immunization with pCCL25 elicits anti-influenza A/PR/8/34 neutralizing antibodies. Serial 4-fold dilutions of individual serum samples from immunized mouse groups are incubated with influenza A/PR/8/34 (100 TCID50 virus per well), and results from the chicken erythrocyte sedimentation assay are shown as the serum dilution at which 2000TCID50/ml of influenza A/PR/8/34 can be neutralized. Data represent individual mouse sera (n = 10) in 6 replicates per dilution for each immunized mouse group. Representative wells of non-neutralized virus (agglutination) are shown for control and pR8HA immunized groups, and a single well depicting neutralized virus (no agglutination or a pool of red blood cells at bottom of well) representing pCCL25 co-immunized group. (C–D) Systemic co-immunization with pCCL25 protects mice from influenza A/PR/8/34 intranasal challenge. Average percent weight loss (morbidity) and death (survival) in groups of mice immunized and challenged were as depicted in the immunization schedule shown in Figure 4A. Vector immunized mice (diamonds) lost weight rapidly (C) and 100% (n = 10) died by day 9 post infection (D). Mice that were immunized with pPR8HA (triangles) alone lost an average 18% body weight (C) by day 10 and 80% died (D) by day 10 post challenge. The two surviving mice in the pPR8HA (triangles) immunized group regained weight and survived. Mice immunized with the CCR9 ligand, pCCL25, had a slight reduction in weight (4.7% weight loss) (squares, (C), but all ultimately survived challenge [squares, in panel (D)]. Using log rank analysis, pVAX vs. pR8HA (p = 0.0005), pVAX vs. CCL25 (p < 0.0001), pR8HA vs. pR8HA/CCL25 (p = 0.0003).](/cms/asset/91eb526e-c149-48eb-8aa2-4dca11eba8a8/khvi_a_10922574_f0005.gif)
Lethal influenza A intranasal challenge.
Three co-immunizations of pCCL25 in combination with pPR8HA were performed on groups of 10 mice each, with each injection separated by 2 weeks (immunization schedule shown in ). After an 8-week rest, the mice were challenged intranasally under anesthesia as described in “Materials and Methods.” After challenge, vector-immunized animals lost up to 30% weight by day 5 and had to be humanely euthanized; thus 100% of these mice succumb to infection by day 9-post challenge (). In addition, mice that were immunized with pPR8HA (triangles) alone lost an average 18% weight by day 9 p.i. (post infection), and only 20% (n = 2) survived by day 12 p.i. The two surviving mice in the pPR8HA immunized group regained weight after day 12 p.i. and survived challenge (triangle symbol). Mice co-immunized with the CCR9 ligand, pCCL25, exhibited weight loss curves similar to the pPR8HA group through day 5, but then recovered rapidly and ultimately survived infection (). They displayed a slight reduction in weight (4.7% weight loss) (squares, ), but all ultimately survived challenge (squares, ). Using log rank analysis gave the following results: pVAX vs. pR8HA (p = 0.0005), pVAX vs. CCL25 (p < 0.0001), pR8HA vs. pR8HA/CCL25 (p = 0.0003). These data further support the ability of the CCR9 ligand adjuvant to provide protective immunity in this challenge system.
Discussion
One important challenge facing the development of effective vaccine platforms, including DNA vaccines, is the generation of efficacious antigen-specific secretory-IgA (S-IgA) antibodies and mucosal CD8+ cytotoxic T lymphocyte immune responses that mediate protection at portals of pathogen entry. Several reports show that the antigenic plasmids need to be delivered at mucosal sites in order to elicit protective mucosal immunity.Citation8,Citation11,Citation36-Citation39 In the study reported here, we demonstrate that the mucosal chemokine, CCL25, can generate protective mucosal immune responses when co-delivered systemically as DNA expression plasmids in combination with pathogen-specific plasmid antigens.
While previous studies have demonstrated that mucosal vaccination is critical for eliciting a mucosal immune response, it has been argued for a considerable period of time that gut and other mucosal sites (lungs, lacrimal glands, genitourinary tract, etc.) comprise a unique mucosal recirculation system separate from the systemic compartment.Citation40 However, ample evidence is available to suggest the concept that cells activated at non-mucosal sites or peripheral sites can acquire the ability to migrate to different mucosal tissues surfaces.Citation41 It has also been reported in both mice and humans that co-delivery of an adjuvant with antigen in the skin induces humoral and cellular responses in the mucosa.Citation42-Citation49 Furthermore, the mucosal responses elicited were protective against mucosal challenge with live virus.Citation42,Citation45
Based on findings that define a role for CCL25/CCR9 interactions in the trafficking of B and T lymphocytes, as well as antigen presenting cells, our data support the notion that through expression of CCL25 at sites outside of mucosal tissues, we could elicit antigen-specific humoral and cellular immune responses at systemic and mucosal sites. Systemic cellular and humoral responses were also augmented as shown by enhanced IFN-λ secretion in the spleen as well as by sera IgG. However, it is important to note that the ratio of IgA to IgG was skewed to a greater degree in mucosal-chemokine immunized mice. CCR9 is expressed on all antigen-specific cells homing to the small intestine and on a fraction of IgA secreting B cells in the small intestine.Citation50 Studies are underway to further understand how ligand/receptor families exert differential effects at mucosal sites and our data support a role for CCR9+/CD11c+ cells in the DLN of immunized mice. In addition to infiltrating DC populations, it is likely that circulating CCR9+ mucosal lymphocytes (about 1%) are being recruited to the immunization site rather than undergoing chemotaxis from mucosal tissues. It is possible that the ectopic expression of the mucosal chemokines in the muscle and corresponding DLNs recruits peripheral circulating mucosal T cells, which are subsequently primed with antigen in the DLNs and then recirculate back to mucosal sites as expression of specific mucosal chemokine wanes. Based upon prior studies it is likely that expression of the plasmid-derived chemokine persists only for a few days.Citation51 The now primed T cells would then be free to re-enter the peripheral circulation and traffic to their preferred mucosal sites. Alternatively, the ectopic chemokine expression may be capable of reprogramming responding peripheral immune cells to upregulate mucosal markers, changing their phenotype to resemble that of a T cell found in the mucosa, causing the antigen-experienced cell to home to mucosal sites. However, this is less likely as it would require prior expression of the chemokine ligand on non-mucosal destined T cells at high enough numbers for these cells to be expanded. In addition, enhanced expression of CCL25 in the DLNs following vaccination would upregulate on CCR9 recently activated T and B cells. Activated antigen-specific cells generated in DLNs could disseminate to GALT, as CCR9 would enable them to traffic to effector mucosal sites. Alternatively, CCR9+ DCs capable of presenting HIV-specific peptides could traffic from DLNs to Peyer’s patches and MLNs and initiate the production of antigen-specific T cells that would subsequently home to the lamina propria of intestine. However, the exact mechanisms underlying the induction of antigen-specific cellular and humoral responses at both the systemic and mucosal level need to be further investigated. It is likely that not all CCR9 positive cells that are recruited by the adjuvants and activated in the systemic compartment will end up in the mucosa. This limitation is due to the secondary requirement of expression of specific mucosal adhesion molecules along with the specific chemokine receptor for mucosal trafficking. Further work concentrating on the subset of cells that traffic in response to these adjuvanted DNA approaches will be informative in this regard. The novel use of mucosal chemokines as DNA vaccine adjuvants has potential implications for the development of vaccination strategies that require mucosal immunity.
Materials and Methods
DNA plasmids.
For the antigenic plasmids, DNA vaccine constructs expressing HIV-1 consensus B gag protein (pHIV-1gag) and influenza A strain PR8 hemagglutinin protein (pPR8HA) were prepared as previously described.Citation3,Citation5,Citation21,Citation52 The cloning of the murine mucosal chemokine CCL25 construct (sequence from Gene Bank, accession number NM009138) into pVAX1 cloning vector (Invitrogen) was performed following generation of the chemokine cDNA from RNA extracted from murine thymus (CCL25), followed by PCR using the following primers that contain flanking restriction sites (EcoR1 and Xho1).
pCCL25 sense.
GCC CCC GAA TTC GCC GCC ACC ATG AAA CTG TGG CTT TTT GCC.
pCCL25 antisense.
ATC GGG CCG AGT TAA TTG TTG GTC TTT CTG GG.
PCR conditions for all of the reactions were as follows: 30 cycles at (94°C for 1 min, 55°C for 1.15 min and 72°C for 1.15 min), followed by a final extension at 72°C for 10 min. The 435 (CCL25) base pair PCR products were respectively ligated into pVAX1 cloning vector following a restriction enzyme digestion using EcoR1 and Xho1 (New England Biolabs) restriction sites. The sequences of the restriction sites were designed into the sequence of the primers and match sites in the multiple cloning region of the vector. All positive clones were verified by sequence analysis.
Plasmid transfection and expression analysis.
Expression levels of the plasmid construct were tested following transient transfection of RD cells (ATCC). Cells were plated in 60 × 15 mm tissue culture dishes at a density of 2.5 × 105 cells per well in complete DMEM plus 10%FBS (Hyclone, GIBCO) and allowed to adhere overnight. The next day cells were transfected with CCL25 plasmid (3 μg/well) using FuGENE 6 transfection reagent (Roche Diagnostics) according to the protocol provided by the manufacturer. After 48 h, cell supernatants were harvested and analyzed for the presence of murine CCL25 protein by commercial ELISA kits (R&D Systems).
In vitro translation assay.
The TNT-T7 coupled transcription/translation reticulocyte lysate system (Promega) and [35S] methionine were used to create labeled CCL25 protein products. pVAX vector alone (negative control) or pVAX vector containing CCL25 was labeled with [35S] methionine and added to the reaction mixture according to the instructions supplied by the manufacturer. The reaction was performed at 30°C for 1 h. Labeled proteins were detected using 5 μg purified monoclonal anti-CCL25 (R&D Systems) at 4°C with rotation overnight in RIPA buffer. Approximately 5 mg of protein G-Sepharose beads (Amersham Biosciences) (50 μL of 100 mg/ml stock) were added to each reaction, and the samples were incubated at 4°C, with rotation, for 2 h. The beads were washed three times with binding buffer containing high salt and bovine serum albumin and finally suspended in 2× sample buffer. The protein complexes were eluted from sepharose beads by boiling for 5 min and were electrophoresed on a 12% SDS-PAGE gel (Cambrex). The gel was fixed and treated with amplifying solution (Amersham Biosciences), and dried for 2 h in a Bio-Rad gel drier. The dried gel was exposed to X-ray film at -80°C overnight and developed using the automatic developer (Kodak).
Plasmid immunization, mouse characteristics and animal care.
The quadriceps muscles of 6 to 8 week old female Balb/c mice (Jackson Laboratory) were injected with plasmid DNA formulations containing combinations of vector backbone (pVAX, Invitrogen), chemokine immune adjuvant plasmids (100 μg) and antigenic plasmids (50 μg of pHIV-1gag or 50 μg of pPR8HA, an HA containing plasmid) as described in the figure legends and in previous publications.Citation3,Citation32,Citation53 Formulations contained 0.25% bupivacaine-HCL (Sigma) in isotonic citrate buffer. Co-administration of various gene plasmids involved mixing the designated DNA plasmids so that each experimental group contained the same concentration of DNA (vector backbone added to make all groups equal in DNA concentrations) before injection in a final volume of 100 μl. All DNA was generated using endotoxin-free Qiagen columns. For animal work performed at the University of Pennsylvania, mice were housed in a temperature-controlled, light-cycled facility and their care was under the guidelines of the NIH and the University of Pennsylvania. Mouse work performed at Drexel University College of Medicine took place in the animal facility, and mice were housed in a temperature-controlled, light-cycled facility, with care as required by the guidelines of the NIH and of Drexel University College of Medicine’s Institutional Animal Care and Use Committee and University Laboratory Animal Resources for the humane use of animals in medical research.
Histology of muscle biopsies and isolation of lymphocytes from the DLNs of immunized animals.
For histology experiments, injected Balb/c mouse quadriceps muscle was excised 7 d after immunization and frozen immediately in OCT medium in a dry ice and methanol bath. Frozen muscles were cut into approximately 1,000 serial sections of 5 μm thickness, air-dried and fixed for 10 min in 100% acetone. Fixed sections were stained with H&E before dehydration, mounting and examination for the presence and extent of cellular inflammatory infiltrates. Acetone-fixed 5 μm sections were treated with 0.5% hydrogen peroxide in phosphate-buffered saline (PBS) for 15 min to quench endogenous peroxidases. The sections were washed with PBS, and free biotin was blocked using the avidin/biotin blocking system (Vector Laboratories). Sections were then incubated with the primary antibodies at room temperature for 1 h. For chemokine receptor detection, monoclonal antibodies to mouse CCR9 were used (AbCam) and were co-stained with secondary antibody labeled with biotin. Isotype control antibodies were also utilized (BD Biosciences, Pharmingen). After incubation, the slides were washed 3 times with PBS and then developed with the Vectastain Elite ABC kit and Vector DAB substrate (Vector Laboratories). After substrate development, the sections were washed in water and counterstained with hematoxylin. Slides were rinsed in water, and coverslips were mounted using histomount (Dako). Immunohistochemistry support was provided by Children’s Hospital of Philadelphia, Department of Pathology, Philadelphia, Pennsylvania. The assessment of infiltrating cells was made randomly and in blinded fashion by at least two separate investigators. The experiment was repeated 3 times. We observed similar results in the tibialis anterior (TA) muscle, a smaller muscle routinely used for immunizations. For experiments that harvested DLN lymphocytes, Balb/c mice were immunized with either 100 μg of empty vector pVax or pCCL25 in the TA muscle. DLNs (popliteal and inguinal) were harvested on day 7 post immunization and treated with collagenase in RPMI plus 5% fetal calf serum (FCS) in a 37°C incubator shaker to release lymphocytes. Dead cells were excluded by staining with Aqua blue LIVE/DEAD stain (Invitrogen) and CCR9+ cells were enumerated in the CD3-CD11c+ compartment. The samples were analyzed on a flow cytometer LSRII (BD Biosciences). The data were analyzed using Flowjo software (Tree Star, Inc.) and frequency and absolute numbers of CD3-CD11c+CCR9+ cells were enumerated.
Method for mouse sacrifice, tissue or sample harvest and cell purification for immune analysis.
At endpoints designated in the legends, the animals were sedated using tribromoethanol and appropriate amounts of blood and fecal pellets (pooled or run individually as designated in the legend) were taken prior to animal sacrifice. Following sacrifice, spleen, lung, MLNs and Peyer’s patches were harvested from each mouse in each experimental group and tissue was either pooled or run individually as designated in the figure legends, into a 15 ml conical tube containing R10 medium (RPMI1640 plus 10% fetal bovine serum, 500 μg/l penicillin and 500 μg/l streptomycin; Gibco, Invitrogen). Peyer’s patches were obtained from the duodenum and ileum (small intestine), pooled and disrupted using scissors, tweezers and steel cloth mesh. The lungs, spleens and MLNs from each experimental group were crushed into a single cell suspension using a Seward Stomacher80, put through a 40-μm cell strainer, washed with medium, pelleted and incubated for 5 to 10 min at room temperature in ACK lysing buffer, resulting in the lysis of red blood cells. All cells were washed, resuspended in medium and counted (cell viability was determined using trypan blue stain) using a hemacytometer.
IFN-λ ELISPOT.
IFN-λ ELISPOT was performed as previously described.Citation3 Briefly, ELISPOT Millipore 96-well plates were coated with an R&D Systems anti-mouse IFN-λ capture antibody and incubated for 24 h at 4°C. A total of 200,000 splenocytes from immunized mice were added to each well of an ELISPOT plate and stimulated at 37°C, 5% CO2, in the presence of R10 (negative control), concanavalin A (positive control), or specific peptide (HIV-1 gag, HA) antigens (10 μg/ml). HIV-1 gag 15-mer peptides spanning the entire HIV-1 gag Consensus Clade B subtype, overlapping by 11 amino acids, were acquired from the AIDS Reagent and Reference Repository (Frederick, MD). Alternatively, cells were stimulated with the CD8+ T cell epitope peptide encoding influenza A/PR/8/34 (H1N1) hemagglutinin (IYSTVASSL amino acids 518–526) synthesized by Immunodiagnostics. Following 24 h of stimulation, the plates were washed and incubated for 24 h at 4°C with biotinylated anti-mouse IFN-λ antibody (R&D Systems). The plates were subsequently washed and streptavidin-alkaline phosphatase (R&D Systems) was added to each well and incubated for 2 h at room temperature. The plate was washed, and 5-bromo-4-chloro-3' indolylphosphate p-toluidine salt (BCIP) and nitro blue tetrazolium chloride (NBT) (R&D Systems) were added to each well. Pooled splenic samples from 4 mice were used in these experiments because previous experiments in our laboratory have determined small variability and consistent responses between Balb/c mice within an experimental group receiving vaccination with pHIV-1gag or pPR8HA constructs.
Flow cytometric analysis of intracellular IFN-λ.
HIV-1 specific T cell responses were determined by intracellular cytokine staining using the Cytofix/Cytoperm Kit and a standard protocol (BD Biosciences). All antibodies were purchased from BD Biosciences. Lymphocytes from the MLNs were harvested from individual mice (n = 4) and purified cells were stimulated in the presence of 1 μl/ml GolgiPlug and GolgiStop with R10 medium and dimethyl sulfoxide (negative control), 50 ng/ml PMA and 1 μg/ml ionomycin (positive control), or HIV-1 consensus Clade B gag peptides for 5 h. Prior to surface staining, cells were incubated with the Violet LIVE/DEAD fixable kit (Invitrogen/Molecular Probes) at 37° for 10 min. The cells were blocked with FcR (CD16/CD32) for 15 min at 4°. Prior to permeabilization/fixation cells were stained with CD4-Alexa700 and CD8-PerCP for 30 min at 4°. CD3-PECy5 and IFN-λ PE-Cy7 were included in the intracellular stain for 45 min at 4°. Data from 50,000 live CD3+ lymphocyte gated events were acquired using an LSRII flow cytometer and analyzed using FlowJo software. Responses from the negative control wells were subtracted from the antigenic stimulations prior to graphing. Data are shown from individual mice as well as the averages for each immunization group (± SEM).
Processing of fecal extract for antibody measurement.
Fecal pellets were collected from vaccinated mice and pooled for each experimental group. Stool was dissolved in the following buffer in a specific weight/volume ratio. One gram of thawed stool was dissolved with 4 ml of PBS pH 7.5 (0.05% Tween 20, soybean trypsin inhibitor (100 μg/ml), EDTA (0.05 M) + phenylmethylsulfonylfluoride at 10 mM. The suspension was incubated for 15 min with frequent vortexing, and sediment was pelleted by centrifugation at 1,200 rpm for 5 min. The fecal supernatant was filtered using a 70-μg strainer and centrifuged again at 16,000 rpm for 15 min. Supernatants were further diluted in 1% bovine serum albumin (BSA) or frozen in 0.1% BSA/0.02% sodium azide.
Analysis of IgG and IgA binding antibodies by ELISA.
ELISA was used to determine HIV-1 gag and influenza A/PR/8/34 hemagglutinin specific IgG and IgA in mouse serum and fecal extract as described.Citation54,Citation55 Mouse blood samples were harvested by retroorbital or submandibular bleeds and fecal pellets were obtained from the cage of mouse groups, and subsequently, both serum and fecal samples were processed from mice (number per group is designated in legends) in each experimental group. For measurement of effector (10–15 d following last immunization) anti-HIVgag and anti-influenza IgG and IgA responses, sera and feces were collected for analysis by ELISA. The data shown are representative of 6 independent experiments. Pooled fecal samples from 4 mice were used in these experiments because previous experiments in our laboratory have determined small variability and consistent responses between Balb/c mice within an experimental group receiving vaccination with pHIV-1gag or pPR8HA constructs. For the influenza challenge study, mouse serum and fecal pellets were obtained from 10 mice per experimental group, processed and run individually in ELISA analysis. Enzyme immunoassay/radioimmunoassay plates (Costar) were coated with 0.5 to 2 μg/ml of recombinant HIV-1IIIB gag p24 or UV-inactivated influenza A/PR/8/34 diluted in PBS (Mediatech) at a final volume of 100 μl per well and incubated overnight at 4°C. Plates were washed with PBS/Tween (0.05% Tween 20) 3 times and blocked against non-specific binding with 200 μl of blocking buffer/diluent (3% BSA in PBS) for 2 h at room temperature. The plates were washed and pooled sera or fecal extract from immunized mice diluted in PBS/1%BSA was added to wells at a final volume of 100 μl. Samples were added in triplicate, at dilutions from 1 to 25 to 1 to 3,200 dilution series to determine endpoint titer [defined as the lowest sample dilution at which measurements for experimental group are equal to vector-immunized mouse sample optical density (OD) values] and incubated at room temperature for 2 h or overnight at 4°C. Bound antibodies were detected with horseradish peroxidase-labeled goat anti-mouse IgG or IgA (Santa Cruz) and developed with substrate TMB H2O2 (SIGMA). The color reaction was stopped with 2N H2SO4, and the absorbance at 450 nm was read in an EL312 Bio-Kinetics microplate reader (Bio-Tek Instruments Inc.). The amount of total IgG or IgA in sera or fecal secretions was calculated by interpolating the ODs on calibration curves, using the DeltaSoft II program (BioMetallics, Inc.).
B cell ELISPOT for measurement of IgA and IgG.
B cell ELISPOT was performed as described previouslyCitation56-Citation59 with some modifications as described below. Immunized mice were sacrificed 1 week following the last immunization and antibody-secreting cells were harvested from Peyer’s patches of the collected mouse small intestine. ELISpot 96-well plates were coated with 2 μg/ml of HIV-IIIB p24 protein or UV-inactivated influenza A/PR/8/34 diluted in PBS (Mediatech) at a final volume of 100 μl per well, and incubated for 24 h at 4°C. The plates were washed and blocked for 2 h with 1% BSA. A total of 500,000 mucosal lymphocytes from the immunized mice were added to each well in triplicate and incubated for 5 h at 37°C, 5% CO2. The plates were washed and 100 μl of a 1:2,000 dilution of anti-mouse IgA-biotin (diluted in PBS/1%BSA) were added and incubated overnight at 4°C. The plates were washed, and 100 μl of 1:100 dilution of streptavidin-alkaline phosphatase (diluted in PBS/1% BSA) was added and incubated for 2 h at room temperature. The plates were washed, and BCIP and NBT substrates were added to each well. The plates were rinsed with distilled water, and dried at room temperature. Spots were counted by an automated ELISpot reader (CTL Limited, Inc.) and by eye. Raw values are determined and multiplied by a factor so that data are represented as ASCs per million IgA secreting B cells.
Virus titer and virus neutralization assay.
The virus neutralization was performed in microtiter plates as described previously.Citation60 The influenza A/PR/8/34 (H1N1) per ml was diluted in PBS, and 100 μl aliquots were dispensed into wells of flat-bottom microtiter plates (105.6 TCID50/ml or 100TCID50 virus per well, 2000TCID50/ml) that contained 50 μl of sera from immunized mice, positive controls and negative control sera dilutions, with 6 to 12 replicates per dilution. After 1 h at 37°C, 50 μl of this mixture was added to a monolayer of MDCK cells, plated at a cell density of 2.5 × 106 cells/ml, in ISCCM–0.1% BSA medium. After 8 h, medium that contained trypsin (32 μg/ml) was added to each well, and the cultures were incubated for an additional 2.5 d. Culture supernatants (25 μl) were tested by hemagglutination assay for presence of virus. Using round-bottom polystyrene microtiter plates, culture supernatants were mixed with 25 μl of CRBCs (1% in PBS), and the pattern of RBC sedimentation was recorded after allowing the plates to incubate for 35 min at room temperature. The neutralization titer is defined as the dilution of serum or antibody sample at which infection with 2000TCID50/ml of influenza A/PR/8/34 is neutralized in 50% of the culture.
HA/flu challenge model.
Using the pPR8HA plasmid cohorts of mice (n = 10) were immunized and then boosted at 2-week intervals with the 33 μg dose antigenic plasmid DNA. CCL25 was co-immunized with the influenza antigenic plasmid (pPR8HA) at a 100 μg dose. Additionally one cohort of mice (n = 10) remained naïve and another cohort (n = 10) received vector backbone control. We have previously found that 10 LD50 influenza A/PR/8/34 in Balb/c mice establishes a lethal infection in 100% of naïve animals, > 80% lethal infection in mice receiving 33 μg pPR8HA and < 10% lethal infection in mice receiving a 50 μg pPR8HA dose. Therefore, for challenge studies, Balb/c mice are immunized with 33 μg of pPR8HA antigenic plasmid. Eight weeks after the last boost, the cohorts were challenged intranasally with the dose of 10 LD50 (2000TCID50) live influenza A/PR/8/34. The animals were temporally anesthetized with anesthetic (tribromoethanol) given by intraperitoneal injection and subsequently handled for influenza A/PR/8/34 infection. 30 μl of dosed virus diluted in PBS was applied to the nares of anesthetized mice resulting in the aspiration of the virus into upper and lower airways. Each day following infection, a decrease in total body weight was measured as a surrogate of morbidity and survival post challenge was noted.
Statistical analysis.
Data are presented as the mean ± SD calculated from triplicate wells of assay using pooled specimens from an experimental group or as ± SEM when specimens from individual animals within an experimental group were kept independent as noted in the legends. Where appropriate, the statistical difference between immunization groups was assessed by using a two-tailed, paired Student’s t test, yielding a specific p value for each experimental group. Comparisons between samples with a p value < 0.05 were considered to be statistically different and therefore significant. Kaplan-Meier survival analysis was utilized to depict results for the influenza challenge study and statistical differences between vaccination groups were determined using a log-rank test.
Acknowledgments
We would like to thank the University of Pennsylvania Department of Pathology Histology Core and Daniel Martinez for their help with immunohistochemistry. Amy Quinn and Carolyn Clarke provided molecular cloning expertise. In addition, we would like to acknowledge Dr. Walter Gerhard, Krystyna Mozdzanowska and Darya Zharikova for their helpful comments and expertise on influenza A/PR/8/34 mouse studies and neutralization assays. We would also like to acknowledge Drs. Rafi Ahmed and Rachael Aubert for their knowledge and guidance to carry out the B cell ELISpot assay. Dr. Jiri Mestecky contributed scientific guidance for the humoral analysis on mucosal tissue and fecal extracts. We would like to acknowledge Pamela Fried and Diana Winters from Drexel University College of Medicine Academic Publishing Services for their editorial, formatting and journal submission expertise. This work is supported by grants funded through the National Institute of Health including an F32AI054152 through NIAIDS (MK), a N01A1154 NIAIDS-HVDDT (DBW), a P01AI071739 (DBW) and a T32-A107632 (KAK).
Disclosure of Potential Conflicts of Interest
The laboratory notes possible commercial conflicts associated with this work, which may include: Wyeth, VGX, BMS, Virxsys, Ichor, Merck, Althea, Aldeveron and possibly others.
References
- Schoenly KA, Weiner DB. Human immunodeficiency virus type 1 vaccine development: recent advances in the cytotoxic T-lymphocyte platform “spotty business”. J Virol 2008; 82:3166 - 80; PMID: 17989174
- Kodihalli S, Kobasa DL, Webster RG. Strategies for inducing protection against avian influenza A virus subtypes with DNA vaccines. Vaccine 2000; 18:2592 - 9; http://dx.doi.org/10.1016/S0264-410X(99)00485-5; PMID: 10775793
- Kutzler MA, Robinson TM, Chattergoon MA, Choo DK, Choo AY, Choe PY, et al. Coimmunization with an optimized IL-15 plasmid results in enhanced function and longevity of CD8 T cells that are partially independent of CD4 T cell help. J Immunol 2005; 175:112 - 23; PMID: 15972637
- Chattergoon MA, Saulino V, Shames JP, Stein J, Montaner LJ, Weiner DB. Co-immunization with plasmid IL-12 generates a strong T-cell memory response in mice. Vaccine 2004; 22:1744 - 50; http://dx.doi.org/10.1016/j.vaccine.2004.01.036; PMID: 15068858
- Chattergoon MA, Kim JJ, Yang JS, Robinson TM, Lee DJ, Dentchev T, et al. Targeted antigen delivery to antigen-presenting cells including dendritic cells by engineered Fas-mediated apoptosis. Nat Biotechnol 2000; 18:974 - 9; http://dx.doi.org/10.1038/79470; PMID: 10973219
- Toka FN, Pack CD, Rouse BT. Molecular adjuvants for mucosal immunity. Immunol Rev 2004; 199:100 - 12; http://dx.doi.org/10.1111/j.0105-2896.2004.0147.x; PMID: 15233729
- Neeson P, Boyer J, Kumar S, Lewis MG, Mattias L, Veazey R, et al. A DNA prime-oral Listeria boost vaccine in rhesus macaques induces a SIV-specific CD8 T cell mucosal response characterized by high levels of alpha4beta7 integrin and an effector memory phenotype. Virology 2006; 354:299 - 315; http://dx.doi.org/10.1016/j.virol.2006.06.036; PMID: 16904153
- Lee S, Gierynska M, Eo SK, Kuklin N, Rouse BT. Influence of DNA encoding cytokines on systemic and mucosal immunity following genetic vaccination against herpes simplex virus. Microbes Infect 2003; 5:571 - 8; http://dx.doi.org/10.1016/S1286-4579(03)00108-4; PMID: 12787733
- Kaneko H, Bednarek I, Wierzbicki A, Kiszka I, Dmochowski M, Wasik TJ, et al. Oral DNA vaccination promotes mucosal and systemic immune responses to HIV envelope glycoprotein. Virology 2000; 267:8 - 16; http://dx.doi.org/10.1006/viro.1999.0093; PMID: 10648178
- Kusakabe K, Xin KQ, Katoh H, Sumino K, Hagiwara E, Kawamoto S, et al. The timing of GM-CSF expression plasmid administration influences the Th1/Th2 response induced by an HIV-1-specific DNA vaccine. J Immunol 2000; 164:3102 - 11; PMID: 10706700
- Biragyn A, Belyakov IM, Chow YH, Dimitrov DS, Berzofsky JA, Kwak LW. DNA vaccines encoding human immunodeficiency virus-1 glycoprotein 120 fusions with proinflammatory chemoattractants induce systemic and mucosal immune responses. Blood 2002; 100:1153 - 9; http://dx.doi.org/10.1182/blood-2002-01-0086; PMID: 12149191
- Bagarazzi ML, Boyer JD, Javadian MA, Chattergoon MA, Shah AR, Cohen AD, et al. Systemic and mucosal immunity is elicited after both intramuscular and intravaginal delivery of human immunodeficiency virus type 1 DNA plasmid vaccines to pregnant chimpanzees. J Infect Dis 1999; 180:1351 - 5; http://dx.doi.org/10.1086/314978; PMID: 10479171
- Wang B, Dang K, Agadjanyan MG, Srikantan V, Li F, Ugen KE, et al. Mucosal immunization with a DNA vaccine induces immune responses against HIV-1 at a mucosal site. Vaccine 1997; 15:821 - 5; http://dx.doi.org/10.1016/S0264-410X(96)00259-9; PMID: 9234524
- Kutzler MA, Kraynyak KA, Nagle SJ, Parkinson RM, Zharikova D, Chattergoon M, et al. Plasmids encoding the mucosal chemokines CCL27 and CCL28 are effective adjuvants in eliciting antigen-specific immunity in vivo. Gene Ther 2010; 17:72 - 82; http://dx.doi.org/10.1038/gt.2009.112; PMID: 19847203
- Kraynyak KA, Kutzler MA, Cisper NJ, Khan AS, Draghia-Akli R, Sardesal NY, et al. Systemic immunization with CCL27/CTACK modulates immune responses at mucosal sites in mice and macaques. Vaccine 2010; 28:1942 - 51; http://dx.doi.org/10.1016/j.vaccine.2009.10.095; PMID: 20188250
- McKenzie BS, Corbett AJ, Johnson S, Brady JL, Pleasance J, Kramer DR, et al. Bypassing luminal barriers, delivery to a gut addressin by parenteral targeting elicits local IgA responses. Int Immunol 2004; 16:1613 - 22; http://dx.doi.org/10.1093/intimm/dxh163; PMID: 15466913
- Bourges D, Zhan Y, Brady JL, Braley H, Caminschi I, Prato S, et al. Targeting the gut vascular endothelium induces gut effector CD8 T cell responses via cross-presentation by dendritic cells. J Immunol 2007; 179:5678 - 85; PMID: 17947639
- Rainone V, Dubois G, Temchura V, Überla K, Clivio A, Nebuloni M, et al. CCL28 induces mucosal homing of HIV-1-specific IgA-secreting plasma cells in mice immunized with HIV-1 virus-like particles. PLoS One 2011; 6:e26979; http://dx.doi.org/10.1371/journal.pone.0026979; PMID: 22066023
- Boyer JD, Kim J, Ugen K, Cohen AD, Ahn L, Schumann K, et al. HIV-1 DNA vaccines and chemokines. Vaccine 1999; 17:Suppl 2 S53 - 64; http://dx.doi.org/10.1016/S0264-410X(99)00235-2; PMID: 10506409
- Sin J, Kim JJ, Pachuk C, Satishchandran C, Weiner DB. DNA vaccines encoding interleukin-8 and RANTES enhance antigen-specific Th1-type CD4(+) T-cell-mediated protective immunity against herpes simplex virus type 2 in vivo. J Virol 2000; 74:11173 - 80; http://dx.doi.org/10.1128/JVI.74.23.11173-11180.2000; PMID: 11070014
- Kim JJ, Yang JS, Dentchev T, Dang K, Weiner DB. Chemokine gene adjuvants can modulate immune responses induced by DNA vaccines. J Interferon Cytokine Res 2000; 20:487 - 98; http://dx.doi.org/10.1089/10799900050023906; PMID: 10841077
- McKay PF, Barouch DH, Santra S, Sumida SM, Jackson SS, Gorgone DA, et al. Recruitment of different subsets of antigen-presenting cells selectively modulates DNA vaccine-elicited CD4+ and CD8+ T lymphocyte responses. Eur J Immunol 2004; 34:1011 - 20; http://dx.doi.org/10.1002/eji.200324840; PMID: 15048711
- Kutzler MA, Weiner DB. Developing DNA vaccines that call to dendritic cells. J Clin Invest 2004; 114:1241 - 4; PMID: 15520855
- Feng N, Jaimes MC, Lazarus NH, Monak D, Zhang C, Butcher EC, et al. Redundant role of chemokines CCL25/TECK and CCL28/MEC in IgA+ plasmablast recruitment to the intestinal lamina propria after rotavirus infection. J Immunol 2006; 176:5749 - 59; PMID: 16670280
- Hieshima K, Kawasaki Y, Hanamoto H, Nakayama T, Nagakubo D, Kanamaru A, et al. CC chemokine ligands 25 and 28 play essential roles in intestinal extravasation of IgA antibody-secreting cells. J Immunol 2004; 173:3668 - 75; PMID: 15356112
- Nakayama T, Hieshima K, Izawa D, Tatsumi Y, Kanamaru A, Yoshie O. Cutting edge: profile of chemokine receptor expression on human plasma cells accounts for their efficient recruitment to target tissues. J Immunol 2003; 170:1136 - 40; PMID: 12538668
- Lazarus NH, Kunkel EJ, Johnston B, Wilson E, Youngman KR, Butcher EC. A common mucosal chemokine (mucosae-associated epithelial chemokine/CCL28) selectively attracts IgA plasmablasts. J Immunol 2003; 170:3799 - 805; PMID: 12646646
- Wurbel MA, Malissen M, Guy-Grand D, Malissen B, Campbell JJ. Impaired accumulation of antigen-specific CD8 lymphocytes in chemokine CCL25-deficient intestinal epithelium and lamina propria. J Immunol 2007; 178:7598 - 606; PMID: 17548595
- Marsal J, Svensson M, Ericsson A, Iranpour AH, Carramolino L, Márquez G, et al. Involvement of CCL25 (TECK) in the generation of the murine small-intestinal CD8alpha alpha+CD3+ intraepithelial lymphocyte compartment. Eur J Immunol 2002; 32:3488 - 97; http://dx.doi.org/10.1002/1521-4141(200212)32:12<3488::AID-IMMU3488>3.0.CO;2-E; PMID: 12442331
- Svensson M, Johansson-Lindbom B, Wurbel MA, Malissen B, Márquez G, Agace W. Selective generation of gut-tropic T cells in gut-associated lymphoid tissues: requirement for GALT dendritic cells and adjuvant. Ann N Y Acad Sci 2004; 1029:405 - 7; http://dx.doi.org/10.1196/annals.1309.025; PMID: 15681793
- Stenstad H, Ericsson A, Johansson-Lindbom B, Svensson M, Marsal J, Mack M, et al. Gut-associated lymphoid tissue-primed CD4+ T cells display CCR9-dependent and -independent homing to the small intestine. Blood 2006; 107:3447 - 54; http://dx.doi.org/10.1182/blood-2005-07-2860; PMID: 16391017
- Sundström P, Lundin SB, Nilsson LA, Quiding-Järbrink M. Human IgA-secreting cells induced by intestinal, but not systemic, immunization respond to CCL25 (TECK) and CCL28 (MEC). Eur J Immunol 2008; 38:3327 - 38; http://dx.doi.org/10.1002/eji.200838506; PMID: 19003934
- Wendland M, Czeloth N, Mach N, Malissen B, Kremmer E, Pabst O, et al. CCR9 is a homing receptor for plasmacytoid dendritic cells to the small intestine. Proc Natl Acad Sci U S A 2007; 104:6347 - 52; http://dx.doi.org/10.1073/pnas.0609180104; PMID: 17404233
- Morteau O, Gerard C, Lu B, Ghiran S, Rits M, Fujiwara Y, et al. An indispensable role for the chemokine receptor CCR10 in IgA antibody-secreting cell accumulation. J Immunol 2008; 181:6309 - 15; PMID: 18941222
- Mata M, Travers PJ, Liu Q, Frankel FR, Paterson Y. The MHC class I-restricted immune response to HIV-gag in BALB/c mice selects a single epitope that does not have a predictable MHC-binding motif and binds to Kd through interactions between a glutamine at P3 and pocket D. J Immunol 1998; 161:2985 - 93; PMID: 9743362
- Toka FN, Rouse BT. Mucosal application of plasmid-encoded IL-15 sustains a highly protective anti-Herpes simplex virus immunity. J Leukoc Biol 2005; 78:178 - 86; http://dx.doi.org/10.1189/jlb.1004621; PMID: 15817700
- Sin JI, Kim JJ, Arnold RL, Shroff KE, McCallus D, Pachuk C, et al. IL-12 gene as a DNA vaccine adjuvant in a herpes mouse model: IL-12 enhances Th1-type CD4+ T cell-mediated protective immunity against herpes simplex virus-2 challenge. J Immunol 1999; 162:2912 - 21; PMID: 10072541
- Okada E, Sasaki S, Ishii N, Aoki I, Yasuda T, Nishioka K, et al. Intranasal immunization of a DNA vaccine with IL-12- and granulocyte-macrophage colony-stimulating factor (GM-CSF)-expressing plasmids in liposomes induces strong mucosal and cell-mediated immune responses against HIV-1 antigens. J Immunol 1997; 159:3638 - 47; PMID: 9317164
- Eo SK, Lee S, Kumaraguru U, Rouse BT. Immunopotentiation of DNA vaccine against herpes simplex virus via co-delivery of plasmid DNA expressing CCR7 ligands. Vaccine 2001; 19:4685 - 93; http://dx.doi.org/10.1016/S0264-410X(01)00241-9; PMID: 11535317
- McDermott MR, Bienenstock J. Evidence for a common mucosal immunologic system. I. Migration of B immunoblasts into intestinal, respiratory, and genital tissues. J Immunol 1979; 122:1892 - 8; PMID: 448111
- Masopust D, Vezys V, Wherry EJ, Barber DL, Ahmed R. Cutting edge: gut microenvironment promotes differentiation of a unique memory CD8 T cell population. J Immunol 2006; 176:2079 - 83; PMID: 16455963
- Belyakov IM, Hammond SA, Ahlers JD, Glenn GM, Berzofsky JA. Transcutaneous immunization induces mucosal CTLs and protective immunity by migration of primed skin dendritic cells. J Clin Invest 2004; 113:998 - 1007; PMID: 15057306
- Berry LJ, Hickey DK, Skelding KA, Bao S, Rendina AM, Hansbro PM, et al. Transcutaneous immunization with combined cholera toxin and CpG adjuvant protects against Chlamydia muridarum genital tract infection. Infect Immun 2004; 72:1019 - 28; http://dx.doi.org/10.1128/IAI.72.2.1019-1028.2004; PMID: 14742549
- Glenn GM, Scharton-Kersten T, Vassell R, Mallett CP, Hale TL, Alving CR. Transcutaneous immunization with cholera toxin protects mice against lethal mucosal toxin challenge. J Immunol 1998; 161:3211 - 4; PMID: 9759833
- Godefroy S, Goestch L, Plotnicky-Gilquin H, Nguyen TN, Schmitt D, Staquet MJ, et al. Immunization onto shaved skin with a bacterial enterotoxin adjuvant protects mice against respiratory syncytial virus (RSV). Vaccine 2003; 21:1665 - 71; http://dx.doi.org/10.1016/S0264-410X(02)00733-8; PMID: 12639488
- Guebre-Xabier M, Hammond SA, Epperson DE, Yu J, Ellingsworth L, Glenn GM. Immunostimulant patch containing heat-labile enterotoxin from Escherichia coli enhances immune responses to injected influenza virus vaccine through activation of skin dendritic cells. J Virol 2003; 77:5218 - 25; http://dx.doi.org/10.1128/JVI.77.9.5218-5225.2003; PMID: 12692224
- Güereña-Burgueño F, Hall ER, Taylor DN, Cassels FJ, Scott DA, Wolf MK, et al. Safety and immunogenicity of a prototype enterotoxigenic Escherichia coli vaccine administered transcutaneously. Infect Immun 2002; 70:1874 - 80; http://dx.doi.org/10.1128/IAI.70.4.1874-1880.2002; PMID: 11895950
- Scharton-Kersten T, Yu J, Vassell R, O’Hagan D, Alving CR, Glenn GM. Transcutaneous immunization with bacterial ADP-ribosylating exotoxins, subunits, and unrelated adjuvants. Infect Immun 2000; 68:5306 - 13; http://dx.doi.org/10.1128/IAI.68.9.5306-5313.2000; PMID: 10948159
- Yu J, Cassels F, Scharton-Kersten T, Hammond SA, Hartman A, Angov E, et al. Transcutaneous immunization using colonization factor and heat-labile enterotoxin induces correlates of protective immunity for enterotoxigenic Escherichia coli. Infect Immun 2002; 70:1056 - 68; http://dx.doi.org/10.1128/IAI.70.3.1056-1068.2002; PMID: 11854183
- Kunkel EJ, Kim CH, Lazarus NH, Vierra MA, Soler D, Bowman EP, et al. CCR10 expression is a common feature of circulating and mucosal epithelial tissue IgA Ab-secreting cells. J Clin Invest 2003; 111:1001 - 10; PMID: 12671049
- Chattergoon MA, Robinson TM, Boyer JD, Weiner DB. Specific immune induction following DNA-based immunization through in vivo transfection and activation of macrophages/antigen-presenting cells. J Immunol 1998; 160:5707 - 18; PMID: 9637479
- Robinson HL, Boyle CA, Feltquate DM, Morin MJ, Santoro JC, Webster RG. DNA immunization for influenza virus: studies using hemagglutinin- and nucleoprotein-expressing DNAs. J Infect Dis 1997; 176:Suppl 1 S50 - 5; http://dx.doi.org/10.1086/514176; PMID: 9240695
- Kim JJ, Nottingham LK, Sin JI, Tsai A, Morrison L, Oh J, et al. CD8 positive T cells influence antigen-specific immune responses through the expression of chemokines. J Clin Invest 1998; 102:1112 - 24; http://dx.doi.org/10.1172/JCI3986; PMID: 9739045
- Ogawa T, Tarkowski A, McGhee ML, Moldoveanu Z, Mestecky J, Hirsch HZ, et al. Analysis of human IgG and IgA subclass antibody-secreting cells from localized chronic inflammatory tissue. J Immunol 1989; 142:1150 - 8; PMID: 2644350
- Mestecky J, Jackson S, Moldoveanu Z, Nesbit LR, Kulhavy R, Prince SJ, et al. Paucity of antigen-specific IgA responses in sera and external secretions of HIV-type 1-infected individuals. AIDS Res Hum Retroviruses 2004; 20:972 - 88; http://dx.doi.org/10.1089/aid.2004.20.972; PMID: 15585085
- Slifka MK, Ahmed R. Limiting dilution analysis of virus-specific memory B cells by an ELISPOT assay. J Immunol Methods 1996; 199:37 - 46; http://dx.doi.org/10.1016/S0022-1759(96)00146-9; PMID: 8960096
- Brown KA, Kriss JA, Moser CA, Wenner WJ, Offit PA. Circulating rotavirus-specific antibody-secreting cells (ASCs) predict the presence of rotavirus-specific ASCs in the human small intestinal lamina propria. J Infect Dis 2000; 182:1039 - 43; http://dx.doi.org/10.1086/315808; PMID: 10979897
- Crotty S, Aubert RD, Glidewell J, Ahmed R. Tracking human antigen-specific memory B cells: a sensitive and generalized ELISPOT system. J Immunol Methods 2004; 286:111 - 22; http://dx.doi.org/10.1016/j.jim.2003.12.015; PMID: 15087226
- Qadri F, Ryan ET, Faruque AS, Ahmed F, Khan AI, Islam MM, et al. Antigen-specific immunoglobulin A antibodies secreted from circulating B cells are an effective marker for recent local immune responses in patients with cholera: comparison to antibody-secreting cell responses and other immunological markers. Infect Immun 2003; 71:4808 - 14; http://dx.doi.org/10.1128/IAI.71.8.4808-4814.2003; PMID: 12874365
- Mozdzanowska K, Feng J, Eid M, Zharikova D, Gerhard W. Enhancement of neutralizing activity of influenza virus-specific antibodies by serum components. Virology 2006; 352:418 - 26; http://dx.doi.org/10.1016/j.virol.2006.05.008; PMID: 16777168