Abstract
Many Canadians received a novel AS03-adjuvanted vaccine during the 2009 influenza A/H1N1 pandemic. Longer term implications of adjuvant use were unclear: would anti-H1N1 immune responses persist at high levels and, if so, could that result in increased or unusual adverse effects upon re-exposure to H1N1pdm09 antigen in the trivalent influenza vaccine (TIV) for 2010–11? To answer these questions, adults given AS03-adjuvanted H1N1pdm09 vaccine (Arepanrix®, GSK Canada) 9–10 mo earlier were enrolled in an evaluator-blinded, crossover trial to receive 2010–2011 non-adjuvanted TIV (Fluviral®, GSK Canada) and placebo 10 d apart, in random order. Adverse effects were monitored for 7 d after each injection. Vaccine-attributable adverse event (VAAE) rates were calculated by subtracting rates after placebo from those after vaccine. Blood was obtained pre-vaccination and 21–30 d afterward to measure hemagglutination inhibiting antibody titers. In total, 326 participants were enrolled and 321 completed the study. VAAE rates were low except for myalgia (18.6%) and injection site pain (63.2%). At baseline, H1N1pdm09 titers ≥ 40 were present in 176/325 subjects (54.2%, 95% confidence interval 48.6, 59.7), with a geometric mean titer (GMT) of 37.4 (95% CI 32.8, 42.6). Post-immunization, 96.0% (95% CI 92.3, 97.8) had H1N1pdm09 titers ≥ 40, with GMT of 167.4 (95% CI 148.7, 188.5). Responses to both influenza A strains in TIV were similar, implying no lasting effect of adjuvant exposure. In summary, titers ≥ 40 persisted in only half the participants 9–10 mo after adjuvanted pandemic vaccine but were restored in nearly all after TIV vaccination, with minimal increase in adverse effects.
Introduction
During the A/H1N1 2009 global pandemic,Citation1 adjuvanted influenza vaccines were widely used for the first time. Along with many other countries, Canada deployed a novel AS03-adjuvanted H1N1pdm09 vaccine (Arepanrix®, GSK Canada).Citation2-Citation5 All Canadians older than 6 mo were eligible to receive this vaccine, which accounted for over 96% of pandemic vaccine doses administered. The adjuvant is an oil-in-water emulsion containing squalene and tocopherol. This vaccine was used in a dose-sparing formulation (3.75 μg hemagglutinin per adult dose), one dose of which was adequately immunogenic in naïve adults.Citation2-Citation5 By the end of the mass campaign in Canada, over 40% of the population had received this vaccine.Citation6 It was not known at the time if adjuvant use would be associated with persistently elevated antibody responses or the rapid decay typically seen after seasonal inactivated vaccines.Citation7 In studies launched while the pandemic vaccine was initially deployed in 2009, evidence of prior infection was already present in one-third of adults under 60 y of age.Citation4,Citation5 These primed individuals developed strong booster antibody responses to H1N1pdm09 vaccination, with greater potential for persistence. Both natural infection and vaccination with adjuvant were expected to elicit greater cell-mediated immune responses than observed following standard trivalent seasonal vaccines.
In 2010 the A/H1N1pdm09 virus continued to circulate with minimal antigenic change. The World Health Organization recommended that the same H1N1pdm09 vaccine strain be included in the trivalent inactivated vaccine (TIV) for the 2010–2011 influenza season.Citation8 Some candidates for re-vaccination with the H1N1pdm09 strain would have been strongly primed by prior infection, vaccination with adjuvant or both. It was certainly conceivable that such individuals might have greater residual cellular immunity and/or specific antibody levels than ordinarily encountered after seasonal influenza vaccination and that this might predispose them to increased vaccine reactions. Vaccination in the presence of specific cellular immunity has been linked to increased injection site reactions after childhood booster doses of adjuvanted, pertussis-containing vaccinesCitation9,Citation10 while vaccination in the presence of high titers of antitoxin increased reaction rates after repeated tetanus toxoid boosters.Citation11 It was not known at the time if residual cellular or humoral immunity to H1N1 would be sufficient to cause reactions with a first booster dose of H1N1 antigen. The post-pandemic situation was unique in modern experience with influenza vaccines and warranted investigation, especially given the potential for unpredictable variations in TIV reactogenicity such as occurred in children in Western Australia in 2010 with a particular TIV formulation.Citation12
We conducted a pre-season study to rapidly assess the safety of re-vaccinationCitation13 with H1N1pdm09 antigen. Eligible adults had received the adjuvanted pandemic vaccine 9–10 mo previously, from community providers. Participants received TIV and placebo one week apart, in random, undisclosed order. Adverse events were documented daily; rates following placebo were subtracted from rates after vaccine to determine vaccine-attributable rates.Citation14,Citation15 The results were used to inform subsequent seasonal vaccination programs in Canada. Secondary objectives were to measure residual antibody titers to H1N1pdm09 virus in previously vaccinated adults and the magnitude of the booster response to re-vaccination with this antigen, in comparison to A/H3N2 responses.
Results
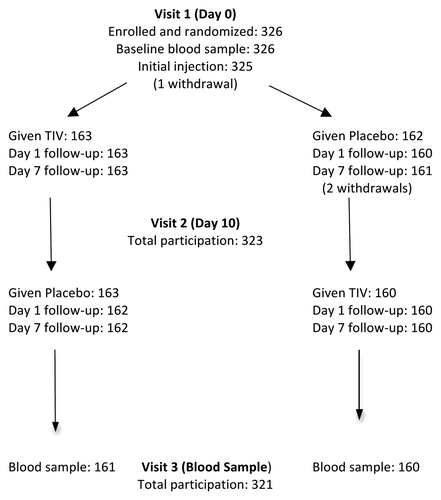
Enrollment totaled 326 subjects, who were predominantly female and Caucasian/white (). Randomized groups were well matched (). The rate of prior seasonal influenza vaccination was high as most subjects were employed by or affiliated with the participating health care institutions. Reported chronic health conditions were generally of minor nature. Centers commenced enrollment on the same day (9 August 2010) and completed it within 5 d. Compliance with the protocol was high () enabling per protocol analysis of safety and immunogenicity data.
Table 1. Characteristics of study participants in a randomized, crossover trial of 2010–11 inactivated, trivalent influenza vaccine vs. placebo
Figure 1. Summary of participation in crossover protocol. Comment: one subject withdrew before the initial injection, a second withdrew shortly after the initial injection (placebo, with code break) and a third withdrew prior to the second injection. Two subjects opted out of visit 3.
Safety observations
As shows, TIV vaccination was associated with higher than background (post-placebo) rates for myalgia, tiredness, headache, malaise, arthralgia, sleep disturbance and diarrhea during the week after vaccination. Only myalgia, tiredness and headache occurred at vaccine-attributable rates (VAR) > 5.0%, with myalgia leading at 18.6%. Most subjects who reported these symptoms rated them as mild/moderate (). The peak VAR’s for myalgia and headache were reached later during the day of vaccination, at 16.9% and 4.9%, respectively, while reports of tiredness peaked next day, with a VAR of 5.8%. Reported rates of myalgia and headache then declined to match those following placebo from day 3 onwards, with tiredness rates normalizing one day later. A similar pattern was seen with reports of malaise, arthralgia, sleep disturbance and diarrhea. Background rates during the week after placebo were > 5% for headache, tiredness, myalgia and malaise (), illustrating the value of controlled observations for this age group.
Table 2. Rates of general symptoms reported by study participants during days 0–6 after masked TIV vaccine or saline placebo injections, including vaccine-attributable rates (as rate difference)
At the injection site, pain was reported by 63.2% of participants (204/323) during the week after TIV vaccine and by 8.0% (26/323) after placebo. TIV-related pain was rated as mild (186 subjects, 57.6%) or moderate (18 subjects, 5.6%) with no instances of severe pain. Local redness followed TIV in 45 participants (13.9%), with 38 (11.8%) having mild (< 25 mm) and 7 (2.2%) having moderate (26–99 mm) redness. Local swelling followed TIV in 31 participants (9.6%), with a single instance (0.3%) of severe swelling (≥ 100 mm). Injection site symptoms were short-lived: all resolved by Day 6 after vaccination, well before second injections (data not shown).
A strong association was evident on day 1 after vaccination between reported rates of injection site pain and myalgia. Among 145 subjects with pain on that day, 41 (28.3%) also had generalized myalgia whereas among 180 subjects without pain, only 10 (5.6%) reported myalgia (p < 0.0001, Chi- square test).
During the first 24 h after TIV vaccination 32 subjects (9.8%) reported 47 new-onset respiratory symptoms. These participants more often reported concurrent myalgia than those without new respiratory symptoms (34.4% vs. 13.6%, p < 0.01, odds ratio 3.3, 95% confidence interval 1.5, 7.4). No significant association was detected with other general symptoms (data not shown). Twenty-one vaccinated subjects reported a single respiratory symptom (sore throat-8, cough-6, hoarseness-3, wheezing-2, red eyes-2), all of which were rated mild. Eleven vaccinated subjects (3.4%) had multiple, new-onset respiratory symptoms (), more consistent with oculorespiratory syndrome (ORS).Citation16,Citation17
Table 3. Participants with multiple new respiratory symptoms within one day after vaccine or placebo administration
All symptoms were rated mild or moderate and none of those affected sought medical attention. During the first 24 h after placebo injection, 11 subjects (3.4%) reported 19 new-onset respiratory symptoms, mainly sore throat (6), hoarseness (3), cough (3), chest tightness (3) and ocular redness (1). The frequency of respiratory symptoms was significantly lower after placebo than after TIV (3.4% vs. 9.8%, p < 0.001) but the spectrum of symptoms was similar in both settings. Only 3 subjects had multiple respiratory symptoms after placebo (), yielding a vaccine-attributable rate of ORS-like symptoms of 2.2% (p < 0.01).
Other notable adverse events included minor allergy-like reactions (generalized pruritus, rash, facial swelling) in 4 vaccinees, none serious or early-onset. Two vaccinees developed neurologic symptoms, with pain or paresthesia radiating down the injected arm. Both instances began within hours after vaccination, lasted several days, were rated severe but resolved fully. Other adverse events after placebo injection included a vasovagal reaction (1), cardiac palpitations (1) and hyperventilation syndrome (1), all beginning within minutes after the injection. Only one subject was hospitalized during the study, after a motor vehicle accident.
Despite treatment blinding, 75% of subjects correctly guessed when they had received TIV, based on Day 7 interviews. A preliminary report of blinded safety data after first injections was presented to the DSMB encompassing all Day 1 and most Day 7 observations. So few severe adverse events had occurred that the DSMB approved continuing as planned. A detailed safety report was presented to the Public Health Agency of Canada 32 d after study commencement (September 10, 2010) and was widely shared with provincial and territorial immunization program administrators before public programs began.
Immunogenicity data
At study entry, HAI antibody to H1N1pdm09 was detectable in most subjects () with half having titers ≥ 40. Age influenced the frequency of residual titers ≥ 40, which ranged from 73.4% (95% CI 60.9,83.7) among 20–29 y olds to 42.5% (95% CI 31.5,57.5) among 40–49 y olds (p = 0.0004, Cochran-Armitage trend test, 2-sided). Following vaccination (), almost all subjects developed titers ≥ 40 and the GMT increased 4.4 fold. A subset of 52 subjects had participated in a trial of AS03-adjuvanted vaccine in 2009.Citation4 One month after the pandemic vaccine, 51 (98.1%) had titers ≥ 40, with a GMT of 285.7 (95% CI 208, 391). Nine months later, 37 (71.2%) retained titers ≥ 40, the GMT having declined 82% to 52.6 (95% CI 39.3, 70.3). After TIV, all but one had titers ≥ 40 and GMT was 180.4 (95% CI 139, 235), a 3.4-fold increase.
Table 4. HAI antibody responses to immunization with 2010 TIV vaccine, in adults previously given adjuvanted H1N1 2009 pandemic vaccine
Responses to A/H3N2 and B components of the vaccine are included in
An association was detected between baseline titers of antibody to A/H1N1pdm09 and post-immunization myalgia. Subjects with baseline titers ≤ 20 were less likely to experience myalgia (25/137, 18%) than those with titers ≥ 80 (31/106, 29%, p < 0.05, odds ratio 0.54, 95%CI 0.298, 0.996). Baseline titers of antibody to H3N2 and B viruses did not show a significant association with myalgia (data not shown). No associations were detected between baseline H1N1 antibody titers and other general symptoms, perhaps because of their infrequency, or with injection site pain or erythema (data not shown).
Discussion
This study provided timely assurance that re-exposure of Canadian adults to H1N1pdm09 antigen contained in TIV for 2010–11 would be safe, following prior vaccination with a novel, adjuvanted pandemic vaccine. The observed rates and severity of common adverse effects were within the ranges previously described for similar TIV vaccines.Citation7,Citation14,Citation15 However, myalgia was reported more frequently in this study than others assessing the same product,Citation14,Citation15 including a similar placebo-controlled, crossover study in 2001–2002Citation14 in which the vaccine-attributable rate of myalgia was 2.6% (95% CI 0.4, 4.8) compared with 18.6% (95% CI 13.2, 24.1) in the present study. Peak rates of myalgia in the current study were as high as 28% in subjects with injection site pain and 29% in those with baseline H1N1pdm09 HAI titers ≥ 80. However, most instances were mild/moderate and short-lived. To our knowledge, such a titer threshold effect for an adverse event following influenza vaccination has not previously been reported. The lack of significant association with baseline titers of H3N2 and B antibodies raises the possibility that the myalgia reported by our subjects was triggered by elements of the immune response specific to the H1N1pdm09 antigen itself, the adjuvant or the combination. The strong association between myalgia and injection site pain suggests that whatever immune mechanisms were involved in the local inflammatory process, they generated cytokines with this adverse effect, such as interferon. However, confirmation of this hypothesis would have required inclusion of a control group of adults not previously exposed to the adjuvanted vaccine. In a study of UK childrenCitation16 similarly revaccinated with TIV in 2010 after receiving 2 doses of AS03-adjuvanted or whole virion H1N1pdm09 vaccine a year earlier, injection site redness and severe local reactions were more frequent in children < 5 y of age given the adjuvanted vaccine, supporting the possibility of unique effects after the latter vaccine. Two studies of Canadian childrenCitation17,Citation18 re-exposed to H1N1pdm09 antigen in 2010 after receiving AS03-adjuvanted vaccine in 2009 reported no unusual increase in adverse effects after vaccination but also lacked a comparison group not primed with adjuvanted vaccine.
Our screening for possible unusual adverse effects of re-vaccination with H1N1/2009 antigen included monitoring for symptoms of oculorespiratory syndrome (ORS).Citation19-Citation22 We expected viral respiratory infections to be infrequent during a mid-summer study. ORS was described in 2000 as an adverse effect of a Canadian-manufactured TIV (Fluviral®, Shire Biologics) for the 2000–2001 season.Citation19,Citation20,Citation22 The syndrome was defined as onset within 2–24 h after vaccination of any of bilateral red eyes, facial swelling, cough, hoarseness, sore throat, difficulty swallowing, wheezing, chest tightness or difficulty breathing, not obviously associated with an allergic reaction or respiratory infection. The overall ORS rate in 2000 was estimated at 3.4% of vaccinated adults, ranging as high as 16% in women 40–59 y old.Citation22 Between 10–24% of individuals with ORS consulted a health care provider.Citation19,Citation20 A re-formulated product for 2001–2002 (using a second virus splitting detergent) was shown to cause mild ORS at a rate of 2.9% among vaccinated adults.Citation14 In the present study we applied a more stringent case definition than used for surveillance of case reports, given the high rate at which vaccinees and controls described new-onset, single, mild respiratory symptoms. With a requirement for two or more eligible symptoms, the observed rate of ORS-like symptoms after vaccination was 3.4%, with a vaccine-attributable rate of 2.5%. All instances were mild to moderate, with none requiring medical attention. On this basis it seemed unlikely that ORS would occur with increased frequency or severity in public programs. While we detected some infrequent events such as generalized pruritus and limb pain/paresthesia it was not possible to predict whether they were unique to the study population or would be seen as infrequent events in the mass programs. Because of their timing and evolution, the neurological events were likely related to the “act of vaccination” rather than the vaccine components themselves, as has been previously reported in other immunization programs.Citation23
Ferguson et al.Citation2 recently reported that 83% of adults 18–65 y of age given one dose of AS03-adjuvanted H1N1pdm09 vaccine in a prospective study had HAI titers ≥ 40 six months after the vaccination. In the present study, 9–10 mo after such vaccination in field conditions, 54% of adults 19–59 y old still had titers ≥ 40. Both studies showed greater titer declines in older than younger adults, as was also described after administration of two doses of MF59-adjuvanted pandemic vaccine.Citation24 This TIV-like decay rateCitation7 likely reflected the low dose of antigen in the AS03-adjuvanted vaccine, intended as a dose-sparing strategy. Rapid loss of protection after this dosage could have been problematic had the pandemic continued so the optimal balance between dose minimization and duration of protection warrants further study. Effective memory responses were evident after the low dose of antigen as all subjects boosted well with TIV 9–10 mo later, with almost 100% developing titers ≥ 40. However, the post-vaccination GMT of H1N1pdm09 antibodies was just 2.7-fold higher than the GMT of H3N2 antibodies, while the GM fold-rise and seroconversion rates were nearly identical with the two influenza A strains, speaking against any long-lived benefit in terms of HAI responses after exposure to adjuvanted vaccine.
A strength of this study was the randomized, blinded, placebo-controlled design. Our experiences with anxiety-related events after placebo injections and with closely-scrutinized respiratory symptoms underscore the value of placebo-controlled observations when assessing vaccine safety. The crossover trial design provides an efficient means of obtaining placebo-controlled observations in perfectly matched groups. Unfortunately, reactogenicity of the vaccine and placebo differed sufficiently to enable 75% of participants to correctly guess when they had been given TIV, potentially diminishing their objectivity. Recruiting mainly healthcare workers may have increased symptom ascertainment bias but facilitated high protocol compliance. Participants had a high rate of past TIV vaccinations, residual effects of which may have narrowed the differences between H1N1pdm09 and H3N2 responses. Other limitations included our inability to include participants known to have had natural H1N1pdm09 infection, to determine if they responded differently to the subsequent seasonal vaccination. As only single pandemic and seasonal influenza vaccines were assessed, our observations may not reflect antibody decline after other H1N1pdm09 vaccines or the effects of re-exposure to other seasonal influenza vaccines for 2010–2011. In Dutch adultsCitation24 given two doses of MF59-adjuvanted H1N1pdm09 vaccine, antibody decline was remarkably similar to our observations, suggesting that the shared components (squalene and H1N1 antigen) determined the pattern of decline. Second doses of the MF59-adjuvanted vaccine increased titers significantly only in persons over age 50 so the dosing regimens were not too dissimilar between that study and the present one.
Methods and Participants
In this prospective, randomized, evaluator-blinded, crossover trial, each subject received vaccine and placebo, in random sequence. The study was conducted at 5 academic centers across Canada belonging to the PHAC/CIHR Influenza Research Network (PCIRN),Citation4,Citation5 during August and September, 2010. Participating centers were located in Quebec City, Montreal, Ottawa, Calgary and Vancouver. A number of strategies were employed to accelerate enrollment and the safety evaluation, which have been described separately.Citation13
Eligible subjects were 20–59 y of age, in generally good health. Stable chronic health conditions without immunocompromise were acceptable. Subjects had to have received AS03-adjuvanted A/H1N1pdm09 influenza vaccine (Arepanrix®) in late 2009. Exclusion criteria included: allergy to egg or other vaccine constituent; bleeding disorder; pregnancy; immune compromise from medication or illness; recent blood product infusion; or prior receipt of a 2010–2011 influenza vaccine. Informed consent was obtained from each subject at study entry. The study was conducted in accordance with the Code of Ethics of the World Medical Association (Declaration of Helsinki) and was approved by the research ethics board of each participating institution. The ClinicalTrials.gov registry identifier was NCT01140009.
A web-based central randomization method (Daciforms®) was used to assign subjects (1:1) to receive influenza vaccine or placebo at the first visit. Randomization assignments were computer-generated (Proc Plan in SAS, SAS Institute) in balanced blocks of four and stratified by sex and age (20–39 and 40–59 y). Participants were given the opposite assignment at Visit 2, ten days later, with masking maintained. The 10-d interval was selected to allow resolution of any adverse effects after vaccination.
The study vaccine (Fluviral®) was an egg-derived, formalin-inactivated, detergent-split preparation, containing thimerosal as preservative, formulated for the 2010–11 Northern Hemisphere winter season. It contained 15 µg of hemagglutinin from each of A/California/7/2009 (H1N1v)-like, A/Perth/16/2009 (H3N2)-like and B/Brisbane/60/2008-like viruses, per 0.5 mL dose. A single lot was used (#AFLLA574AB), obtained prior to commercial sale authorization with approval of the Biologic and Genetic Therapies Directorate of Health Canada. The placebo was normal saline for injection, locally sourced. Injectables were prepared out of sight of subjects; vaccine and placebo were identical in appearance. Both were stored at 2−8°C and injected into the deltoid muscle using 25 gauge needles, one inch (25 mm) long. Only the nurse who administered injections had access to the treatment information; all other study staff remained blinded, including safety interviewers.
Participants were observed for at least 15 min after each injection. During this time they were given instructions about using a daily symptom diary (supplied) to record any solicited or unsolicited local, respiratory or general symptoms, including fever, for 6 d after each injection. An electronic thermometer was supplied to measure oral temperature, as was a device to measure injection site redness and swelling. Subjects were contacted by telephone one and seven days after each injection to review and record any symptoms. Day 7 interviews included a question about which injection (vaccine or placebo) subjects thought that they had received, with blinding maintained. The purpose of the initial telephone contact was early detection of any severe symptoms. Health care utilization and symptom resolution were reviewed during each clinic visit and throughout the period of study participation. Safety information was entered into a secure, web-based reporting system (Daciforms®) within 24 h after each encounter and reviewed centrally for completeness. A study statistician tallied severe adverse event numbers daily and cumulatively, without breaking the assignment code, to monitor for any increased vaccine reactogenicity. A Data Safety Monitoring Board (DSMB) reviewed severe adverse event rates after the first round of injections, before the second round was undertaken.
Blood samples (8–10 mL) were obtained from subjects at study entry and 21–30 d after the TIV vaccination. To determine the correct sampling date it was necessary to un-blind the group assignment after safety assessments were completed 7 d after the second injection. Blood samples were processed promptly and sera were stored at -20°C or colder pending testing at the national reference laboratory. Serum pairs were tested concurrently, in duplicate, using hemagglutination inhibition (HAI) assays for each vaccine strain per WHO methods.Citation25 Live viruses were used for influenza A assays and ether-treated viruses for influenza B assays. Minimum sensitivity was a titer of 1:10. Results of duplicate tests on samples were expressed as the geometric mean titer (GMT).
Adverse event rates were determined daily and cumulatively for days 0–6 after each injection. The cumulative rate of individual symptoms after placebo was subtracted from the rate after vaccination to determine the vaccine-attributable rate. Symptom severity ratings were provided by subjects according to criteria printed on the diary forms. Severe symptom rates were the primary safety outcome, defined as fever > 40.0°C, injection site redness or swelling ≥ 100 mm diameter or any symptom that precluded normal daily activities or prompted medical attention. Serologic responses were analyzed according to standard international (EMEA/CHMP)Citation26 criteria for adults < 60 y old given seasonal influenza vaccine. Seroprotection was considered the primary serologic outcome measure, defined as an HAI titer ≥ 40. Geometric mean titers were calculated, with any test-negative sample assigned to value of 5. Geometric mean fold rise was calculated as the within-subjects ratios of the post-vaccination HAI titer to the baseline HAI titer. The seroconversion rate was defined as either a 4-fold or greater titer rise or conversion from a negative baseline titer to one ≥ 40.
A sample size of 300 evaluable subjects was desired to permit detection of severe or unusual adverse events at rates as low as 1% with 90% probability. Event rate differences after vaccine and placebo ≥ 6% could be detected with 80% probability (α = 0.05) with 300 observations after each treatment, using Fisher’s exact test. To allow for drop-outs, the intended enrollment was 320 subjects, 64 per center.
Conclusion
The data from this study showed that adults given a low-dose, ASO3-adjuvanted H1N1pdm09 vaccine in 2009 had substantially reduced rates of seroprotection (HAI titers ≥ 40) 9–10 mo later. While the low-dose formulation was intended to be dose-sparing, the short duration of protection could have been problematic had the pandemic been more prolonged. However, individuals given a subsequent non-adjuvanted dose of TIV containing the H1N1pdm09 antigen boosted well, reflecting excellent immune memory. The second vaccination with the H1N1pdm09 antigen was generally well tolerated and did not generate unusually high anti-H1N1 titers. These observations may prove useful in designing dosing regimens for future adjuvanted influenza vaccines.
| Abbreviations: | ||
| BMI | = | body mass index |
| CHMP | = | Committee for Medicinal Products for Human Use |
| CI | = | confidence interval |
| DSMB | = | Data safety monitoring board |
| EMEA | = | European Medicines Evaluation Agency |
| GMT | = | geometric mean titer |
| HAI | = | hemagglutination inhibition |
| H1N1pdm09 | = | swine-derived A/H1N1 virus responsible for the 2009 pandemic |
| ORS | = | oculorespiratory syndrome |
| PCIRN | = | Public Health Agency of Canada/Canadian Institutes of Health Influenza Research Network |
| SD | = | standard deviation |
| TIV | = | trivalent inactivated influenza vaccine |
| VAAE | = | vaccine-attributable adverse event |
| WHO | = | World Health Organization |
Acknowledgments
All named authors participated in the implementation of the study, including contributions toward the study design and gathering and interpretation of the data. All authors were involved in drafting the article and approved the final draft.
Contributing PCIRN Influenza Research Network investigators included Gaston De Serres MD, PhD (Institut national de santé publique du Québec, Laval University), James Kellner MD (University of Calgary, Alberta Children’s Hospital), Simon Dobson MD (University of British Columbia, BC Children’s Hospital), Nathalie Bastien PhD (National Microbiology Laboratory) and Barbara Law MD (Public Health Agency of Canada).
We thank the DSMB members: Gaston De Serres MD (chair), Harold Rode MD, Robert Pless MD (Public Health Agency of Canada) and Jean Luc Grenier MD (Direction de la santé publique des Laurentides).
We greatly appreciated the skilled assistance of the project manager (Carol LaJeunesse), data manager (Kim Marty), statistician (Shuyu Fan) and the coordinators and staff at each participating center. Presented in brief at the Fourteenth Annual Conference on Vaccine Research in Baltimore, MD, on May 16–18, 2011 (Abstract S18).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Financial Disclosure Statement
This study was funded entirely by a grant to the Public Health Agency of Canada/Canadian Institutes of Health Research Influenza Research Network (PCIRN). GSK Canada supplied the influenza vaccine (Fluviral) for this trial and contributed an unrestricted grant toward a similar, concurrent PCIRN trial in children (ref. Citation18), with investigators that included D.W.S., O.G.V., B.J.W., Y.L. and S.A.H. D.W.S. was the principal investigator and has received previous study funds and consulting honoraria from GSK, sanofi, Novartis and Pfizer.
Trademark Statement
Arepanrix and Fluviral are trademarks of the GlaxoSmithKline group of companies.
Related Research Data
References
- Girard MP, Tam JS, Assossou OM, Kieny MP. The 2009 A (H1N1) influenza virus pandemic: A review. Vaccine 2010; 28:4895 - 902; http://dx.doi.org/10.1016/j.vaccine.2010.05.031; PMID: 20553769
- Ferguson M, Risi G, Davis M, Sheldon E, Baron M, Li P, et al. Safety and long-term humoral immune response in adults after vaccination with an H1N1 2009 pandemic influenza vaccine with or without AS03 adjuvant. J Infect Dis 2012; 205:733 - 44; http://dx.doi.org/10.1093/infdis/jir641; PMID: 22315336
- Roman F, Vaman T, Gerlach B, Markendorf A, Gillard P, Devaster JM. Immunogenicity and safety in adults of one dose of influenza A H1N1v 2009 vaccine formulated with and without AS03A-adjuvant: preliminary report of an observer-blind, randomised trial. Vaccine 2010; 28:1740 - 5; http://dx.doi.org/10.1016/j.vaccine.2009.12.014; PMID: 20034605
- Scheifele DW, Ward BJ, Dionne M, Vanderkooi O, Loeb M, Coleman BL, et al. Compatibility of AS03-adjuvanted A/H1N1pdm09 and seasonal trivalent influenza vaccines in adults: results of a randomized, controlled trial. Vaccine In press
- Rubinstein E, Predy G, Sauvé L, Hammond GW, Aoki F, Sikora C, et al. The responses of Aboriginal Canadians to adjuvanted pandemic (H1N1) 2009 influenza vaccine. CMAJ 2011; 183:E1033 - 7; http://dx.doi.org/10.1503/cmaj.110196; PMID: 21788422
- Canadian community health survey: H1N1 vaccinations. Ottawa (ON): Statistics Canada; 2010. Available: www.statcan.gc.ca/daily-quotidien/100719/dq100719b-eng.htm (accessed 2011 Sept 12).
- Beyer WEP, Nauta JJP, Palache AM, Giezeman KM, Osterhaus ADME. Immunogenicity and safety of inactivated influenza vaccines in primed populations: a systematic literature review and meta-analysis. Vaccine 2011; 29:5785 - 92; http://dx.doi.org/10.1016/j.vaccine.2011.05.040; PMID: 21624411
- World Health Organization. Recommended viruses for influenza vaccines for use in the 2010-2011 northern hemisphere influenza season. Available: www.who.int/influenza/vaccines/recommendations/recommendation2010_11north/en/index/html (accessed 2011 Sept 12).
- Scheifele DW, Ochnio JJ, Halperin SA. Cellular immunity as a potential cause of local reactions to booster vaccination with diphtheria and tetanus toxoids and acellular pertussis antigens. Pediatr Infect Dis J 2009; 28:985 - 9; http://dx.doi.org/10.1097/INF.0b013e3181a9cc2a; PMID: 19755930
- Rowe J, Yerkovich ST, Richmond P, Suriyaarachchi D, Fisher E, Feddema L, et al. Th2-associated local reactions to the acellular diphtheria-tetanus-pertussis vaccine in 4- to 6-year-old children. Infect Immun 2005; 73:8130 - 5; http://dx.doi.org/10.1128/IAI.73.12.8130-8135.2005; PMID: 16299307
- Levine L, Edsall G. Tetanus toxoid: what determines reaction proneness?. J Infect Dis 1981; 144:376; http://dx.doi.org/10.1093/infdis/144.4.376; PMID: 7288216
- Armstrong PK, Dowse GK, Effler PV, Carcione D, Blyth CC, Richmond PC, et al. Epidemiological study of severe febrile reactions in young children in Western Australia caused by a 2010 trivalent inactivated influenza vaccine. BMJ Open 2011; 1:e000016; http://dx.doi.org/10.1136/bmjopen-2010-000016; PMID: 22021725
- Scheifele DW, Marty K, LaJeunesse C, Fan SY, Bjornson G, Langley JM, et al. Strategies for successful rapid trials of influenza vaccine. Clin Trials 2011; 8:699 - 704; http://dx.doi.org/10.1177/1740774511419868; PMID: 21900340
- Scheifele DW, Duval B, Russell ML, Warrington R, DeSerres G, Skowronski DM, et al. Ocular and respiratory symptoms attributable to inactivated split influenza vaccine: evidence from a controlled trial involving adults. Clin Infect Dis 2003; 36:850 - 7; http://dx.doi.org/10.1086/368189; PMID: 12652385
- Jackson LA, Gaglani MJ, Keyserling HL, Balser J, Bouveret N, Fries L, et al. Safety, efficacy, and immunogenicity of an inactivated influenza vaccine in healthy adults: a randomized, placebo-controlled trial over two influenza seasons. BMC Infect Dis 2010; 10:71; http://dx.doi.org/10.1186/1471-2334-10-71; PMID: 20236548
- Walker WT, de Whalley P, Andrews N, Oeser C, Casey M, Michaelis L, et al. H1N1 antibody persistence 1 year after immunization with an adjuvanted or whole-virion pandemic vaccine and immunogenicity and reactogenicity of subsequent seasonal influenza vaccine: a multicenter follow-on study. Clin Infect Dis 2012; 54:661 - 9; http://dx.doi.org/10.1093/cid/cir905; PMID: 22267719
- Gilca V, De Serres G, Hamelin ME, Boivin G, Ouakki M, Boulianne N, et al. Antibody persistence and response to 2010-2011 trivalent influenza vaccine one year after a single dose of 2009 AS03-adjuvanted pandemic H1N1 vaccine in children. Vaccine 2011; 30:35 - 41; http://dx.doi.org/10.1016/j.vaccine.2011.10.062; PMID: 22063386
- Langley JM, Scheifele DW, Quach C, Vanderkooi O, Ward B, McNeil S, et al. Safety and immunogenicity of 2010-2011 H1N1 2009-containing trivalent inactivated influenza vaccine in children 12-59 months of age previously given AS03-adjuvanted H1N1 2009 pandemic vaccine: a PHAC/CIHR Influenza Research Network (PCIRN) study. Vaccine 2012; 23:3389 - 94; http://dx.doi.org/10.1016/j.vaccine.2012.03.046
- De Serres G, Grenier JL, Toth E, Ménard S, Roussel R, Tremblay M, et al. The clinical spectrum of the oculo-respiratory syndrome after influenza vaccination. Vaccine 2003; 21:2354 - 61; http://dx.doi.org/10.1016/S0264-410X(03)00094-X; PMID: 12744866
- Skowronski DM, Strauss B, De Serres G, MacDonald D, Marion SA, Naus M, et al. Oculo-respiratory syndrome: a new influenza vaccine-associated adverse event?. Clin Infect Dis 2003; 36:705 - 13; http://dx.doi.org/10.1086/367667; PMID: 12627354
- De Serres G, Toth E, Ménard S, Grenier JL, Roussel R, Tremblay M, et al. Oculo-respiratory syndrome after influenza vaccination: trends over four influenza seasons. Vaccine 2005; 23:3726 - 32; http://dx.doi.org/10.1016/j.vaccine.2005.01.154; PMID: 15882534
- Boulianne N, De Serres G, Duval B, Shadmani R, Rochette L. Clinical manifestations and incidence of oculo-respiratory syndrome following influenza vaccination--Quebec, 2000. Can Commun Dis Rep 2001; 27:85 - 90; PMID: 11396047
- Sever JL, Brenner AI, Gale AD, Lyle JM, Moulton LH, West DJ, Anthrax Vaccine Export Committee. Safety of anthrax vaccine: a review by the Anthrax Vaccine Expert Committee (AVEC) of adverse events reported to the Vaccine Adverse Event Reporting System (VAERS). Pharmacoepidemiol Drug Saf 2002; 11:189 - 202; http://dx.doi.org/10.1002/pds.712; PMID: 12051118
- Huijskens E, Rossen J, Mulder P, van Beek R, van Vugt H, Verbakel J, et al. Immunogenicity, boostability, and sustainability of the immune response after vaccination against Influenza A virus (H1N1) 2009 in a healthy population. Clin Vaccine Immunol 2011; 18:1401 - 5; http://dx.doi.org/10.1128/CVI.05046-11; PMID: 21795459
- World Health Organization. WHO Manual on Animal Influenza Diagnosis and Surveillance, 2002. Available at: www.who.int/vaccine research/diseases/influenza/WHO manual on animal-diagnosis and surveillance 2002 5.pdf [accessed February 15, 2012]
- Wood JM, Newman RW, Ploss K. The use of correlates of immunity in European Union licensing of influenza vaccines. Dev Biol (Basel) 2003; 115:9 - 16; PMID: 15088770