Abstract
The signal transducer and activator of transcription (STAT) proteins are latent transcription factors that have been shown to be involved in cell proliferation, development, apoptosis, and autophagy. STAT proteins undergo activation by phosphorylation at tyrosine 701 and serine 727 where they translocate to the nucleus to regulate gene expression. STAT1 has been shown to be involved in promoting apoptotic cell death in response to cardiac ischemia/reperfusion and has recently been shown by our laboratory to be involved in negatively regulating autophagy. These processes are thought to promote cell death and restrict cell survival leading to the generation of an infarct. Here we present data that shows STAT1 localizes to the mitochondria and co-immunoprecipitates with LC3. Furthermore, electron microscopy studies also reveal mitochondria from ex vivo I/R treated hearts of STAT1KO mice contained within a double membrane autophagosome indicating that STAT1 may be involved in negatively regulating mitophagy. This is the first description of STAT1 being localized to the mitochondria and also having a role in mitophagy.
Introduction
The signal transducer and activator of transcription (STAT) proteins are a family of transcription factors which have been shown to be involved in many cellular functions including tumorigenesis, apoptosis, cell proliferation, development, and autophagy.Citation1-Citation10 Following activation, STAT1 and 3 are tyrosine phosphorylated and dimerize after which they are translocated to the nucleus. During nuclear translocation, they are phosphorylated at a serine residue located in the C-terminal transactivation domain which is thought to enhance transcriptional activity of the complex.Citation11
Cardiac ischemia/reperfusion (I/R) injury results from the occlusion of blood flow through the coronary arteries leading to cell death in the ventricle of the heart by apoptosis and necrosis which leads to a decreased cardiac output and can result in heart failure.Citation12,Citation13 STAT1 and 3 have been shown to be closely linked during cardiac I/R injury showing opposing mechanisms of action while STAT3 has been shown to be located in the mitochondria and is thought to be involved in modulating the mitochondrial permeability transition pore.Citation14 STAT1, specifically the C-terminus, has been shown to be involved in promoting apoptotic cell death leading to increased infarct size.Citation15-Citation17 The activation of STAT1 during I/R injury is likely to be augmented by the generation of free-radicals from uncoupling of oxidative phosphorylation at the mitochondria. Indeed, data from our laboratory using an in vivo rat model of I/R injury showed that free-radical scavenging using the drug 4-hydroxytempol, showed marked decrease in infarct size with concomitant inhibition of STAT1 phosphorylation.Citation18 Further ex vivo studies using STAT1 knockout animals subjected to I/R injury also showed reduced infarct size further suggesting that STAT1 was an important protein for apoptotic cell death.Citation19 Inhibition of STAT1 phosphorylation using the polyphenolic, green tea extract epigallocatechin-3-gallate in both in vitro and ex vivo studies revealed increased cardiomyocyte survival leading to improved hemodynamic ventricular function and hemodynamic recovery.Citation20
Autophagy is a catabolic process that involves enveloping cellular material with a double membrane vesicle (the autophagosome) in order to generate ATP.Citation21 The formation of the autophagosome begins with the mammalian target of rapamycin (mTOR) which modulates the initiator protein Ulk1.Citation22 The Bcl-2 and Bcl-XL proteins inhibit nucleation by binding to Beclin-1, which becomes phosphorylated by DAP-Kinase resulting in the association of ATG-14, VPS-34, and VPS-15 with Beclin-1, which leads to the association and activation of elongation and maturation of the autophagosome.Citation23-Citation25 The signaling involved in elongation and maturation is complex, but ultimately leads to the lipidation of microtubule-associated protein 1 light chain 3 (MAP-LC3) from LC3-I to LC3-II. Kabeya and colleagues showed that LC3-II appeared to associate with the autophagosome membrane and shares 28% homology with yeast Apg8/Aug7p which has been shown to be important in yeast autophagosome formation.Citation26,Citation27
Studies have shown that during I/R injury, autophagy is activated and is thought to promote cell survival and reduce infarct size. Studies from our laboratory have shown that STAT1, as well as promoting apoptosis, negatively regulates autophagy.Citation19 We propose that by enhancing apoptosis and negatively regulating autophagy, STAT1 promotes the generation of larger infarcts which can lead to heart failure. Recently, another catabolic cellular process, mitophagy, has received much attention.
Mitochondria constitute around 30% of cardiomyocyte cell volume and supply the contracting heart with the ATP required for sustained contraction.Citation28 However, the onset of reperfusion results in the generation of highly reactive oxygen free radicals, or superoxide, which are responsible for much of the cellular damage during injury and which have been shown to induce necrosis and apoptosis.Citation29,Citation30 It was initially thought that autophagy was a non-selective process; however, recent data suggests that autophagy selectively degrades cellular material.Citation28 Indeed, to limit the damage inflicted by oxidative-phosphorylation uncoupling, damaged mitochondria are selectively sequestered for degradation by mitophagy or mitochondrial autophagy which may contribute to cell survival and ultimately improved cardiac function.Citation31 Here we show for the first time by immunofluorescence and subcellular fractionation, that STAT1 is localized to the mitochondria following I/R injury and using co-immunoprecipitation we further show that STAT1 binds to the autophagic marker, LC3. Taken together, we present data which suggests that STAT1 may be involved in regulating the selective degradation of mitochondria by the autophagic pathway or mitophagy.
Results
STAT1 localizes to the mitochondria following simulated ischemia/reperfusion injury
Previous reports have shown that STAT3 localizes to the mitochondrial membrane where it is thought to be involved in the modulation of oxidative-phosphorylation.Citation32 Since STAT1 and 3 often have antagonistic effects we postulated that STAT1 may also reside in/at the mitochondria. Here we show for the first time that STAT1 localizes to the mitochondria following simulated ischemia/reperfusion injury. Under control conditions, it could be seen that STAT1 localized to the mitochondria, however, following simulated I/R injury this localization appeared more punctate ().
Figure 1. Primary cardiomyocytes were seeded onto gelatin coated-glass coverslips and subjected to normoxic (control) conditions or simulated ischemia/reperfusion injury, stained with 25 nM Mitotracker-Red then fixed with 4% paraformaldehyde. Immunofluorescent staining indicated that STAT1 co-localized to the mitochondria before I/R injury but formed more localized, punctate staining following I/R injury (A) while STAT3, formed localized punctate staining both before and after I/R injury (B). In order to confirm the presence of STAT1 in the mitochondrial fraction, subcellular fractionation of primary cardiac fibroblasts under normoxic conditions was performed and the mitochondrial (M), cytosolic (C) and nuclear (N) fractions run on 10% PAGE gels and western blotting indicated the presence of STAT1 in all three fractions (C). In order to ensure that the presence of STAT1 was not due to contamination, the blots were interrogated for the cytosolic marker, GAPDH and the mitochondrial membrane marker, CoxIV (C).
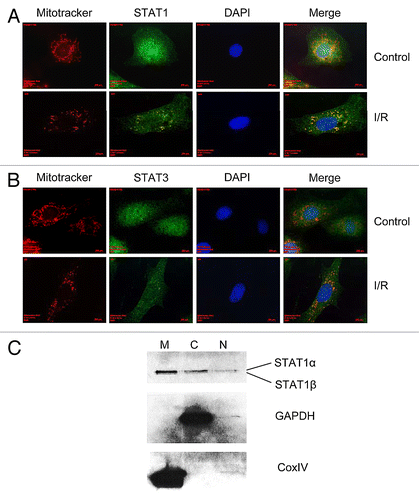
Primary cardiac myocytes were cultured under normoxic conditions for 48 h following isolation, and the media was then replaced with ischemic buffer and the cells subjected to four hours of simulated ischemia and 24 h of reperfusion. Mitochondria were stained with Mitotracker-Red (Molecular Probes) and the cells fixed with 4% (w/v) paraformaldehyde for 15 min then stained with anti-STAT1 or anti-STAT3 antibodies. As shown in , STAT1 was found to form punctate staining where STAT1 co-localized with mitochondria. We found that under the conditions described here, STAT1 mitochondrial localization was more obvious than STAT3 mitochondrial localization ().
To further analyze the presence of STAT1 at the mitochondria, we performed subcellular fractionation studies on primary cardiac fibroblasts under normoxic conditions. Fibroblasts from the primary neonate isolation were cultured in DMEM containing 10% FBS until 90% confluent and then subjected to subcellular fractionation. The mitochondrial, nuclear and cytosolic fractions were run on 10% denaturing PAGE gels and western blotting performed. Here we found a strong presence of STAT1 in the mitochondrial and cytosolic fractions but only a small amount in the nuclear fraction. In order to confirm that the presence of STAT1 was not due to contamination, we performed western blotting for the mitochondrial membrane marker Cox IV and the cytosolic marker glyceraldehyde-6-phosphate dehydrogenase (GAPDH). As shown in , no contamination of GAPDH was seen in the mitochondrial fraction and no Cox IV contamination was found in the mitochondrial fraction. These data, together with the immunofluorescence results suggest that STAT1 is localized to the mitochondria.
Enhanced mitophagy occurs in STAT1−/− mice subjected to ex vivo ischemia/reperfusion injury
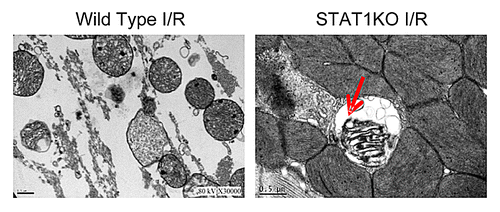
STAT1−/− and wild-type littermate controls were subjected to ex vivo Langendorff perfusion as previously described.Citation19 Using electron microscopy we found that in STAT1−/− animals showed increased numbers of mitochondria enveloped in double membrane structures suggesting that mitochondria were undergoing selective degradation by mitophagy (). These data suggested that STAT1 may be a negative regulator of autophagy since the depletion of STAT1 resulted in increased mitochondrial turnover.
Figure 2. Hearts from STAT1−/− mice and wild type litter mate controls were subjected to ex vivo I/R injury using Langendorff perfusion and then sections subjected to electron microscopy. It was found that cells from STAT1−/− mice had increased numbers of mitochondria (red arrow) located within double membrane structures suggesting they had an increased rate of autophagy.
STAT1 and STAT3 contain conserved LC3-interacting motifs
The LC3-interacting region (LIR) is a consensus binding motif for the autophagosome forming protein LC3 although other binding motifs have been described.Citation33-Citation37 Using protein alignments of STAT1 and STAT3 in frog, fish, mouse, rat, and human, we found a LIR consensus site that was 100% conserved between all species (). Furthermore, we found additional binding motifs which have been described elsewhere further suggesting that STAT1/3 bind to LC3b.Citation37 These motifs were also 100% conserved with the exception of one which was conserved in all species except the frog ().
Figure 3. Protein alignment of STAT1 and STAT3 sequences with LC3-interacting regions (LIR) sequences highlighted. Three LIR sequences showed 100% conservation between STAT1 and STAT1 in human, rat, mouse, frog, and fish (A). The LIR motif YPDI was conserved between STAT1 and STAT3 in all species except in frog (A). LC3 was immunoprecipitated from whole cell lysates of cardiomyocytes, run on 10% PAGE gels and western blot filters interrogated for STAT1 and STAT3 (B). It was found that STAT1 and STAT-3 co-immunoprecipitated with LC3 while the pre-immune showed no presence of STAT1 or STAT-3 suggesting that the immunoprecipitation was specific (B). Primary cardiac fibroblasts were subjected to either control (normoxic) conditions or simulated ischemia/reperfusion injury and the mitochondrial and cytosolic fractions isolated. Western blotting for STAT1/3 revealed STAT1/3 was present in the mitochondrial fraction under control conditions and increased in the cytosolic fraction following I/R injury (C). Only a small amount of contamination from the cytosolic fraction was observed as shown by GAPDH while no mitochondrial contamination was seen in the cytosolic fraction as indicated by CoxIV.
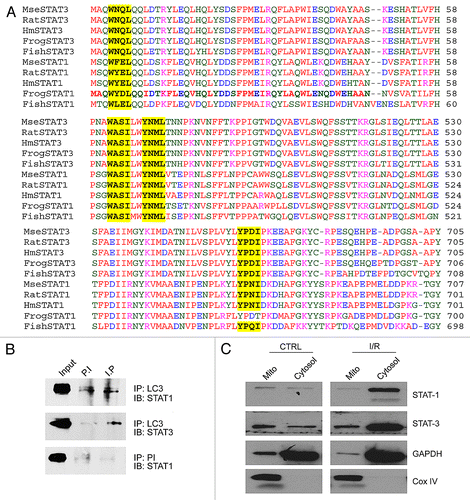
The alignments generated above suggested that STAT1 and STAT3 were likely candidates for binding to LC3. In order to investigate this, we performed co-immunoprecipitation experiments and as shown in , STAT1 co-immunoprecipitated LC3b, probably using the LIR motif identified in the bioinformatics screen in . We further investigated whether STAT3 co-immunoprecipitated with LC3b and found that STAT3 did co-precipitate ().
We next wanted to investigate how STAT1 and STAT3 behaved following simulated ischemia/reperfusion injury. Primary cardiac fibroblasts were subjected to simulated I/R injury as described above and the mitochondrial and cytosolic fractions isolated. Western blotting of these fractions and interrogated for STAT1 and STAT3 revealed that both STAT1 and STAT3 appeared to move from the mitochondrial fraction into the cytosolic fraction following simulated I/R injury ().
Discussion
It has been known for quite some time that autophagy occurs in the heart and has been shown in vitro and in ex vivo.Citation38-Citation40 The first description of mitophagy in the heart was by Decker and Wildenthal, although the mechanisms of mitophagy still remain unclear.Citation39 We recently reported that STAT1 was involved in negatively regulating autophagy as seen in the Langendorff perfused STAT1−/− heart.Citation19 Our electron microscopy data revealed increased numbers of autophagic bodies containing damaged mitochondria in the STAT1−/− animals compared with their wild-type littermate controls. These data led us to believe that, in fact, STAT1 may also be responsible for regulating mitophagy.
Mitochondria are responsible for generating the large concentrations of ATP required for sustained heart contraction.Citation28 However, during I/R injury oxidative phosphorylation becomes uncoupled and highly active superoxide anions begin to “leak” causing damage to cellular proteins and organelles.Citation41,Citation42 Studies have shown that the re-establishment of blood flow leads to additional free-radical generation which ultimately leads to further damage.Citation13 We and others have shown that scavenging free radicals using compounds such as 4-hydroxy-tempol, during the reperfusion phase of injury results in an increase in cell survival as shown by decreased infarct size and inhibition of STAT1 phosphorylation.Citation18,Citation43
The role of autophagy by the catabolism of cellular material to generate ATP for cell survival has been known for quite some time, as has the process of mitophagy.Citation44,Citation45 However, it was initially thought that mitophagy was an almost accidental/non-selective process.Citation28 Recently, it has been suggested that the process of mitophagy may indeed be a very deliberate process in order to remove damaged mitochondria and facilitate cell survival.Citation28 Indeed, two different pathways are involved in the induction of mitophagy.Citation36,Citation46,Citation47 Recent data has shown the involvement of PTEN-inducing putative kinase-1 (PINK1) and the E3-ubiquitin ligase, Parkin, in modulating mitophagy and PINK1 and Parkin mutant cells were also found to have defective mitophagy.Citation48 Currently, it is thought that PINK1 phosphorylates Parkin which is capable of polyubiquitinating VDAC1 which, in turn, recruits p62. P62 recruitment binds to ATG8/LC3b at the autophagosome, thereby targeting the mitochondria to the autophagic pathway.Citation45 These data indicate that the pathway for mitochondrial removal by autophagy is indeed very complex and so the identification of further proteins involved in this process will enable us to not only identify how various cell death/survival pathways are interlinked, but importantly allow us to identify multiple protein targets which can be exploited for therapy.
In this article we present data suggesting that not only does STAT1 appear to localize to the mitochondria but it also seems to bind an LC3b binding partner further suggesting a role for STAT1 in regulating mitophagy. What remains to be elucidated, is the cellular mechanism as to how STAT1 does this. The signaling pathway of autophagy is quite complex and so further studies are required to elucidate how STAT1 is involved in regulating other proteins to fine tune the autophagic/mitophagic response.
Since STAT1 is a pro-apoptotic protein it is thought that it negatively regulates autophagy to facilitate the onset of cell death by inhibiting cell survival mechanisms. Similarly, by inhibiting mitophagy, the clearance of damaged mitochondria is also inhibited further tipping the balance of cell fate in favor of death. This idea is supported by increased numbers of mitochondria located in the autophagosomes of STAT1−/− hearts and further by the observation of STAT1 localizing to the mitochondria and translocating to the cytosolic fraction following simulated I/R injury. This cytosolic translocation of STAT1 would be consistent with it becoming localized in autophagosomes.
The accumulation of damaged mitochondria following injury is thought to lead to local inflammation and ROS signaling resulting in cell death.Citation13 The inhibition of STAT1 may therefore present as a potential target for therapy by increasing the rate of mitophagy thereby eliminating the accumulation of damaged mitochondria and limiting the inflammatory response. Recently, Gottlieb and colleagues suggested that dysregulation of mitophagy/autophagy may contribute to primary pathologies such as I/R injury and pressure-overload.Citation49 Indeed, release of mitochondrial DNA (mtDNA) into the cell is thought to activate the NLRP3 inflammasome leading to IL-1β and IL-18 production.Citation50
In the clinical setting, it has been proposed that the inhibition of STAT1 may be of benefit in order to reduce the infarct area size following myocardial infarction. Since STAT1 has been found to negatively regulate autophagy, it would therefore be advantageous to inhibit its action to allow the clearance of damaged mitochondria thereby facilitating cell survival.
Materials and Methods
Primary neonatal rat ventricular cardiomyocyte isolation
Primary cardiomyocytes were isolated as previously described.Citation17 Animals were housed under SPF conditions and sacrificed by schedule 1 killing in accordance to Home Office guidelines (Animals for Scientific Procedure Act 1986). All isolation buffers were oxygenated by bubbling medical grade oxygen through the solution for 5 min prior to use. Neonate rat pups (Sprague-Dawley) that were <2 d postpartum were decapitated and rinsed in ethanol. The hearts were removed by cutting along the sternum and dissecting the heart through the chest wall. Hearts were washed in isolation buffer (116 mM NaCl, 20 mM HEPES, 0.77 mM nNaH2PO, 5.5 mM Glucose, 5.4 mM KCl, 0.4 mM MgSO containing 600 µg/ml Collagenase type II and 250 µg/ml pancreatin) and cut into small 2 mm pieces then incubated at 37 °C in isolation buffer for 15 min with occasional agitation and the supernatant spun at 1000× rpm for 5 min. The cell pellet was resuspended in 4 ml of fetal bovine serum and kept at 37 °C until plating. The digestion procedure was repeated 7 times and the pellets pooled and pre-plated for 1 h in 15% (v/v) FBS in DMEM to remove contaminating fibroblasts. Cardiomyocytes were then seeded at 2 × 106 cells per well of a 6 well plate which had been pre-coated with 2% (w/v) gelatin. The DMEM containing 15% (v/v) FBS was replaced the following day with maintenance media (DMEM containing 1% [v/v] FBS). Fibroblasts obtained from the pre-plating procedure were cultured in FBS containing 10% (v/v) DMEM until 80–90% before using in downstream experiments.
Simulated ischemia/reperfusion injury
Cell culture media was replaced with ischemic buffer (137 mM NaCl, 12 mM KCl, 0.49 mM MgCl2, 0.9 mM CaCl2·H2O, 4 mM HEPES, 20 mM sodium lactate, and 10 mM deoxyglucose [pH 6.2]) and cells transferred to an ischemic chamber pre-warmed to 37 °C. Simulated ischemia was achieved by addition of 5% CO2 in balanced argon to exclude any oxygen. Cells were subjected to simulated ischemic injury for 4 h after which ischemic buffer was replaced with DMEM containing 1% (v/v) FBS and cultured in 5% CO2 in air (reperfusion) for the indicated times.
Ex vivo ischemia/reperfusion injury
STAT1 knockout (KO) animals were purchased from Taconic Farms Inc. and housed under SPF conditions. STAT1 KO animals (129/Sv) were backcrossed onto a C57B/6 background before used in experiments. STAT1 heterozygotes were paired to generate KO and wild-type littermate controls which were used for the ex vivo studies. Wild-type and STAT1 KO hearts from 10 week old male mice were removed and perfused as previously described.Citation51 Following 20 min of stabilization, hearts were exposed to either control or ischemic conditions, then reperfused for 45 min. Infarct size was assessed using triphenyl tetrazolium chloride staining and computerised planimetry (Planimetry, NIH Image 1.63 software package; National Institutes of Health).
Electron microscopy
Electron microscopy was performed on tissue fixed and embedded as previously described with minimal modifications.Citation52
Immunofluorescence
Cells were grown on UV irradiated coverslips and fixed in 4% (w/v) paraformaldehyde in phosphate buffered saline for 15 min at room temperature. Cells were permeabilized in phosphate buffered saline containing 0.1% (v/v) Triton-X100 for 10 min and blocked in 5% (w/v) bovine serum albumin in PBS for 30 min. Fixed and permeabilized cells were then interrogated with rabbit anti-STAT1 (Santa Cruz), mouse anti-STAT1 (Cell Signaling Technology), mouse anti-STAT-3 (Cell Signaling Technology), and/or mouse anti-LC3b (Abcam) in PBS containing 2.5% (w/v) BSA at a 1/1000 dilution for 1 h at room temperature. Cells were washed three times in PBS and then incubated with Alexa-Fluor secondary antibodies (emission wavelengths Alexa488, Alexa546, Molecular Probes). Mitochondria were stained with Mitotracker Red (emission 588 nm, Molecular Probes) and cell nuclei counter stained with DAPI. Coverslips were mounted onto glass slides using fluorescence mounting media (DAKO) and images acquired using a Zeiss Axioscope inverted fluorescence microscope (Zeiss).
Subcellular fractionation
Subcellular fractionation of mitochondrial and cytosolic fractions was performed using the Qproteome mitochondrial isolation kit (Qiagen). Briefly, cells were washed in 0.9% (w/v) sodium chloride and incubated at 4 °C with rotation for 10 min. Cell lysates were spun at 1000× g for 10 min and the cytosolic fraction stored at −80 °C. The pellet was resuspended in disruption buffer and spun at 1000× g and the retained supernatant spun at 6000× g to pellet the mitochondria. The supernatant was discarded and the pellet resuspended in mitochondria storage buffer then layered with mitochondrial purification buffer. The mitochondrial suspension was spun at 14 000× g and the interphase layer containing the mitochondria was removed. The purified mitochondria were recovered by addition of mitochondrial storage buffer and centrifugation at 8000× g to pellet the mitochondria. The supernatant was removed and the mitochondrial pellet was stored at −80 °C until used in downstream experiments.
Co-immunoprecipitation
Cells were lysed in RIPA buffer (20 mM TRIS-HCl [pH 7.5], 105 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% IGEPAL, 1% deoxycholate, 250 mM sodium pyrophosphate and Complete-mini protease inhibitor cocktail [Roche]) for 5 min on ice and spun at 13 000× rpm for 5 min and the clarified supernatant transferred to a new tube. The protein lysate was pre-cleared with protein A/G sepherose (GE Healthcare) for 1 h at 4 °C, spun at 3000× rpm to pellet the beads and the pre-cleared supernatant transferred to a new tube. Ten percent of the lysate was removed for the input control, and 3 ug of target antibody added and incubated overnight at 4 °C. Proteins were precipitated by addition of protein A/G sepherose and incubated at 4 °C for 1 h with rotation. The protein bound beads were then washed 4 times with ice-cold RIPA buffer and then prepared for western blotting.
Western blotting
Cells were lysed in 1× RIPA buffer (20 mM TRIS-HCl [pH 7.5], 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% [v/v] IGEPAL, 1% [w/v] deoxycholate, 250 mM sodium pyrophosphate and Complete-mini protease inhibitor cocktail [Roche]) and incubated on ice for 15 min. Lysates were passed through a 25 gauge needle 5 times and then spun at 13 000 rpm for 5 min to pellet cell debris. The clarified supernatant was transferred to a clean tube and assayed for protein using the BCA assay kit according to the manufacturer’s instructions (Pierce). Twenty micrograms total protein was run on denaturing PAGE gels in protein running buffer (25 mM TRIS-HCl, 192 mM Glycine, 0.1% [w/v] sodium dodecyl sulfate) and wet-transferred in transfer buffer (25 mM TRIS-HCl, 192 mM Glycine, 0.1% [w/v] sodium dodecyl sulfate containing 20% [v/v] methanol) to Hybond-C nitrocellulose membrane (GE Healthcare). Membranes were blocked for 60 min in 5% (w/v) non-fat dry milk in PBS before being interrogated with detection antibodies. Primary antibody, rabbit-anti-STAT1 were used at 1/1000 dilution in 5% (w/v) non-fat dry milk in PBS for 1 h then washed 3 times in PBS containing 0.1% (v/v) Tween20. Horseradish-peroxidase conjugated secondary antibodies (DAKO) were used at 1/5000 dilution in 5% (w/v) non-fat dry milk for 1 h at room temperature then washed 3 times in PBS containing 0.1% (v/v) Tween20. Bands were visualized using enhanced chemiluminescence (GE Healthcare).
Acknowledgments
The authors would like to thank the British Heart Foundation for funding the project (PG/08/091/26002).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Schindler C. STATs as activators of apoptosis. Trends Cell Biol 1998; 8:97 - 8; http://dx.doi.org/10.1016/S0962-8924(98)01233-1; PMID: 9695817
- Takeda K, Noguchi K, Shi W, Tanaka T, Matsumoto M, Yoshida N, et al. Targeted disruption of the mouse Stat3 gene leads to early embryonic lethality. Proc Natl Acad Sci U S A 1997; 94:3801 - 4; http://dx.doi.org/10.1073/pnas.94.8.3801; PMID: 9108058
- Chang YP, Tsai CC, Huang WC, Wang CY, Chen CL, Lin YS, et al. Autophagy facilitates IFN-gamma-induced Jak2-STAT1 activation and cellular inflammation. J Biol Chem 2010; 285:28715 - 22; http://dx.doi.org/10.1074/jbc.M110.133355; PMID: 20592027
- Coffer P, Lutticken C, van Puijenbroek A, Klop-de Jonge M, Horn F, Kruijer W. Transcriptional regulation of the junB promoter: analysis of STAT-mediated signal transduction. Oncogene 1995; 10:985 - 94; PMID: 7898939
- Harrison DA, Binari R, Nahreini TS, Gilman M, Perrimon N. Activation of a Drosophila Janus kinase (JAK) causes hematopoietic neoplasia and developmental defects. EMBO J 1995; 14:2857 - 65; PMID: 7796812
- Luo H, Hanratty WP, Dearolf CR. An amino acid substitution in the Drosophila hopTum-l Jak kinase causes leukemia-like hematopoietic defects. EMBO J 1995; 14:1412 - 20; PMID: 7729418
- Schwaller J, Frantsve J, Aster J, Williams IR, Tomasson MH, Ross TS, et al. Transformation of hematopoietic cell lines to growth-factor independence and induction of a fatal myelo- and lymphoproliferative disease in mice by retrovirally transduced TEL/JAK2 fusion genes. EMBO J 1998; 17:5321 - 33; http://dx.doi.org/10.1093/emboj/17.18.5321; PMID: 9736611
- Takemoto S, Mulloy JC, Cereseto A, Migone TS, Patel BK, Matsuoka M, et al. Proliferation of adult T cell leukemia/lymphoma cells is associated with the constitutive activation of JAK/STAT proteins. Proc Natl Acad Sci U S A 1997; 94:13897 - 902; http://dx.doi.org/10.1073/pnas.94.25.13897; PMID: 9391124
- Turkson J, Bowman T, Garcia R, Caldenhoven E, De Groot RP, Jove R. Stat3 activation by Src induces specific gene regulation and is required for cell transformation. Mol Cell Biol 1998; 18:2545 - 52; PMID: 9566874
- Yu CL, Meyer DJ, Campbell GS, Larner AC, Carter-Su C, Schwartz J, et al. Enhanced DNA-binding activity of a Stat3-related protein in cells transformed by the Src oncoprotein. Science 1995; 269:81 - 3; http://dx.doi.org/10.1126/science.7541555; PMID: 7541555
- Stark GR, Darnell JE Jr.. The JAK-STAT pathway at twenty. Immunity 2012; 36:503 - 14; http://dx.doi.org/10.1016/j.immuni.2012.03.013; PMID: 22520844
- Maxwell SR, Lip GY. Reperfusion injury: a review of the pathophysiology, clinical manifestations and therapeutic options. Int J Cardiol 1997; 58:95 - 117; http://dx.doi.org/10.1016/S0167-5273(96)02854-9; PMID: 9049675
- Eltzschig HK, Eckle T. Ischemia and reperfusion--from mechanism to translation. Nat Med 2011; 17:1391 - 401; http://dx.doi.org/10.1038/nm.2507; PMID: 22064429
- Boengler K, Hilfiker-Kleiner D, Heusch G, Schulz R. Inhibition of permeability transition pore opening by mitochondrial STAT3 and its role in myocardial ischemia/reperfusion. Basic Res Cardiol 2010; 105:771 - 85; http://dx.doi.org/10.1007/s00395-010-0124-1; PMID: 20960209
- Barry SP, Townsend PA, McCormick J, Knight RA, Scarabelli TM, Latchman DS, et al. STAT3 deletion sensitizes cells to oxidative stress. Biochem Biophys Res Commun 2009; 385:324 - 9; http://dx.doi.org/10.1016/j.bbrc.2009.05.051; PMID: 19450559
- Janjua S, Stephanou A, Latchman DS. The C-terminal activation domain of the STAT-1 transcription factor is necessary and sufficient for stress-induced apoptosis. Cell Death Differ 2002; 9:1140 - 6; http://dx.doi.org/10.1038/sj.cdd.4401082; PMID: 12232802
- Stephanou A, Brar BK, Scarabelli TM, Jonassen AK, Yellon DM, Marber MS, et al. Ischemia-induced STAT-1 expression and activation play a critical role in cardiomyocyte apoptosis. J Biol Chem 2000; 275:10002 - 8; http://dx.doi.org/10.1074/jbc.275.14.10002; PMID: 10744676
- McCormick J, Barry SP, Sivarajah A, Stefanutti G, Townsend PA, Lawrence KM, et al. Free radical scavenging inhibits STAT phosphorylation following in vivo ischemia/reperfusion injury. FASEB J 2006; 20:2115 - 7; http://dx.doi.org/10.1096/fj.06-6188fje; PMID: 16935931
- McCormick J, Suleman N, Scarabelli TM, Knight RA, Latchman DS, Stephanou A. STAT1 deficiency in the heart protects against myocardial infarction by enhancing autophagy. J Cell Mol Med 2012; 16:386 - 93; http://dx.doi.org/10.1111/j.1582-4934.2011.01323.x; PMID: 21447043
- Townsend PA, Scarabelli TM, Pasini E, Gitti G, Menegazzi M, Suzuki H, et al. Epigallocatechin-3-gallate inhibits STAT-1 activation and protects cardiac myocytes from ischemia/reperfusion-induced apoptosis. FASEB J 2004; 18:1621 - 3; PMID: 15319365
- Levine B. Eating oneself and uninvited guests: autophagy-related pathways in cellular defense. Cell 2005; 120:159 - 62; PMID: 15680321
- McCormick J, Knight RA, Barry SP, Scarabelli TM, Abounit K, Latchman DS, et al. Autophagy in the stress-induced myocardium. [Elite Ed] Front Biosci (Elite Ed) 2012; 4:2131 - 41; PMID: 22202025
- Inbal B, Bialik S, Sabanay I, Shani G, Kimchi A. DAP kinase and DRP-1 mediate membrane blebbing and the formation of autophagic vesicles during programmed cell death. J Cell Biol 2002; 157:455 - 68; http://dx.doi.org/10.1083/jcb.200109094; PMID: 11980920
- Zalckvar E, Berissi H, Eisenstein M, Kimchi A. Phosphorylation of Beclin 1 by DAP-kinase promotes autophagy by weakening its interactions with Bcl-2 and Bcl-XL. Autophagy 2009; 5:720 - 2; http://dx.doi.org/10.4161/auto.5.5.8625; PMID: 19395874
- Zalckvar E, Berissi H, Mizrachy L, Idelchuk Y, Koren I, Eisenstein M, et al. DAP-kinase-mediated phosphorylation on the BH3 domain of beclin 1 promotes dissociation of beclin 1 from Bcl-XL and induction of autophagy. EMBO Rep 2009; 10:285 - 92; http://dx.doi.org/10.1038/embor.2008.246; PMID: 19180116
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 2000; 19:5720 - 8; http://dx.doi.org/10.1093/emboj/19.21.5720; PMID: 11060023
- Kirisako T, Baba M, Ishihara N, Miyazawa K, Ohsumi M, Yoshimori T, et al. Formation process of autophagosome is traced with Apg8/Aut7p in yeast. J Cell Biol 1999; 147:435 - 46; http://dx.doi.org/10.1083/jcb.147.2.435; PMID: 10525546
- Kubli DA, Gustafsson AB. Mitochondria and mitophagy: the yin and yang of cell death control. Circ Res 2012; 111:1208 - 21; http://dx.doi.org/10.1161/CIRCRESAHA.112.265819; PMID: 23065344
- Flaherty JT. Myocardial injury mediated by oxygen free radicals. Am J Med 1991; 91:3C 79S - 85S; http://dx.doi.org/10.1016/0002-9343(91)90288-9; PMID: 1928216
- Braunwald E, Kloner RA. Myocardial reperfusion: a double-edged sword?. J Clin Invest 1985; 76:1713 - 9; http://dx.doi.org/10.1172/JCI112160; PMID: 4056048
- Quinsay MN, Thomas RL, Lee Y, Gustafsson AB. Bnip3-mediated mitochondrial autophagy is independent of the mitochondrial permeability transition pore. Autophagy 2010; 6:855 - 62; http://dx.doi.org/10.4161/auto.6.7.13005; PMID: 20668412
- Wegrzyn J, Potla R, Chwae YJ, Sepuri NB, Zhang Q, Koeck T, et al. Function of mitochondrial Stat3 in cellular respiration. Science 2009; 323:793 - 7; http://dx.doi.org/10.1126/science.1164551; PMID: 19131594
- Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy 2011; 7:279 - 96; http://dx.doi.org/10.4161/auto.7.3.14487; PMID: 21189453
- Ichimura Y, Kumanomidou T, Sou YS, Mizushima T, Ezaki J, Ueno T, et al. Structural basis for sorting mechanism of p62 in selective autophagy. J Biol Chem 2008; 283:22847 - 57; http://dx.doi.org/10.1074/jbc.M802182200; PMID: 18524774
- Noda NN, Kumeta H, Nakatogawa H, Satoo K, Adachi W, Ishii J, et al. Structural basis of target recognition by Atg8/LC3 during selective autophagy. Genes Cells 2008; 13:1211 - 8; http://dx.doi.org/10.1111/j.1365-2443.2008.01238.x; PMID: 19021777
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 2007; 282:24131 - 45; http://dx.doi.org/10.1074/jbc.M702824200; PMID: 17580304
- von Muhlinen N, Akutsu M, Ravenhill BJ, Foeglein A, Bloor S, Rutherford TJ, et al. LC3C, bound selectively by a noncanonical LIR motif in NDP52, is required for antibacterial autophagy. Mol Cell 2012; 48:329 - 42; http://dx.doi.org/10.1016/j.molcel.2012.08.024; PMID: 23022382
- Sybers HD, Ingwall J, DeLuca M. Autophagy in cardiac myocytes. Recent Adv Stud Cardiac Struct Metab 1976; 12:453 - 63; PMID: 1032000
- Decker RS, Wildenthal K. Lysosomal alterations in hypoxic and reoxygenated hearts. I. Ultrastructural and cytochemical changes. Am J Pathol 1980; 98:425 - 44; PMID: 7355988
- Decker RS, Poole AR, Crie JS, Dingle JT, Wildenthal K. Lysosomal alterations in hypoxic and reoxygenated hearts. II. Immunohistochemical and biochemical changes in cathepsin D. Am J Pathol 1980; 98:445 - 56; PMID: 6986784
- Ambrosio G, Zweier JL, Duilio C, Kuppusamy P, Santoro G, Elia PP, et al. Evidence that mitochondrial respiration is a source of potentially toxic oxygen free radicals in intact rabbit hearts subjected to ischemia and reflow. J Biol Chem 1993; 268:18532 - 41; PMID: 8395507
- Kim JS, Jin Y, Lemasters JJ. Reactive oxygen species, but not Ca2+ overloading, trigger pH- and mitochondrial permeability transition-dependent death of adult rat myocytes after ischemia-reperfusion. Am J Physiol Heart Circ Physiol 2006; 290:H2024 - 34; http://dx.doi.org/10.1152/ajpheart.00683.2005; PMID: 16399872
- Menegazzi M, Tedeschi E, Dussin D, De Prati AC, Cavalieri E, Mariotto S, et al. Anti-interferon gamma action of epigallocatechin-3-gallate mediated by specific inhibition of STAT1 activation. FASEB J 2001; 15:1309 - 11; PMID: 11344123
- Ernster L, Schatz G. Mitochondria: a historical review. J Cell Biol 1981; 91:227s - 55s; http://dx.doi.org/10.1083/jcb.91.3.227s; PMID: 7033239
- Novak I. Mitophagy: a complex mechanism of mitochondrial removal. Antioxid Redox Signal 2012; 17:794 - 802; http://dx.doi.org/10.1089/ars.2011.4407; PMID: 22077334
- Kirkin V, Lamark T, Sou YS, Bjørkøy G, Nunn JL, Bruun JA, et al. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol Cell 2009; 33:505 - 16; http://dx.doi.org/10.1016/j.molcel.2009.01.020; PMID: 19250911
- Strappazzon F, Vietri-Rudan M, Campello S, Nazio F, Florenzano F, Fimia GM, et al. Mitochondrial BCL-2 inhibits AMBRA1-induced autophagy. EMBO J 2011; 30:1195 - 208; http://dx.doi.org/10.1038/emboj.2011.49; PMID: 21358617
- Kubli DA, Zhang X, Lee Y, Hanna RA, Quinsay MN, Nguyen CK, et al. Parkin protein deficiency exacerbates cardiac injury and reduces survival following myocardial infarction. J Biol Chem 2013; 288:915 - 26; http://dx.doi.org/10.1074/jbc.M112.411363; PMID: 23152496
- Gottlieb RA, Mentzer RM Jr.. Autophagy: an affair of the heart. Heart Fail Rev 2012; In press http://dx.doi.org/10.1007/s10741-012-9367-2; PMID: 23188163
- Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011; 469:221 - 5; http://dx.doi.org/10.1038/nature09663; PMID: 21124315
- Smith RM, Suleman N, Lacerda L, Opie LH, Akira S, Chien KR, et al. Genetic depletion of cardiac myocyte STAT-3 abolishes classical preconditioning. Cardiovasc Res 2004; 63:611 - 6; http://dx.doi.org/10.1016/j.cardiores.2004.06.019; PMID: 15306216
- Scarabelli T, Stephanou A, Rayment N, Pasini E, Comini L, Curello S, et al. Apoptosis of endothelial cells precedes myocyte cell apoptosis in ischemia/reperfusion injury. Circulation 2001; 104:253 - 6; http://dx.doi.org/10.1161/01.CIR.104.3.253; PMID: 11457740