Abstract
A wide variety of full-size monoclonal antibodies (mAbs) and therapeutics derived from alternative antibody formats can be produced through genetic and biological engineering techniques. These molecules are now filling the preclinical and clinical pipelines of every major pharmaceutical company and many biotechnology firms. Metrics for the development of antibody therapeutics, including averages for the number of candidates entering clinical study and development phase lengths for mAbs approved in the United States, were derived from analysis of a dataset of over 600 therapeutic mAbs that entered clinical study sponsored, at least in part, by commercial firms. The results presented provide an overview of the field and context for the evaluation of on-going and prospective mAb development programs. The expansion of therapeutic antibody use through supplemental marketing approvals and the increase in the study of therapeutics derived from alternative antibody formats are discussed.
Monoclonal antibodies (mAbs) are a notably versatile class of therapeutics. Through biological engineering, a wide variety of full-size mAbs and alternative antibody formats can be produced and molecules such as these are now filling the preclinical and clinical pipelines of every major pharmaceutical company and many biotechnology firms. The attraction is partially due to the fact that there are established development and approval pathways and a variety of reliable production methods for the molecules. A total of 28 mAb therapeutic products have been approved by the United States Food and Drug Administration (US FDA) and four are undergoing regulatory review (). Antibody therapeutics also have higher approval success rates and similar development phase lengths compared with those of small molecule drugs.Citation1–Citation4 As evidenced by global sales that exceeded US $1 bn in 2009 for each of at least nine mAb products, physicians and patients have accepted mAbs despite the fact that the products are administered via injection.
The development of therapeutic products is not an easy task. Companies are required by the FDA to establish the safety, efficacy and quality of candidates before marketing approvals are granted, but the pathway to approval cannot be exactly defined at the outset of any program. In order to successfully navigate obstacles, companies must engage in strategic planning of individual programs, as well as their entire product portfolio. Metrics are used by companies to guide them in the planning process; these measures allow companies to compare their own performance to that of the industry as a whole and provide an assessment of efficiency. Industry metrics used for this purpose include the number of novel candidates that enter clinical studies and clinical development and approval phase lengths.
Tufts Center for the Study of Drug Development has periodically reported on trends in the commercial development of therapeutic mAbs.Citation1–Citation3 Metrics for antibody therapeutics development presented here provide context for the evaluation of on-going and prospective mAb development programs and a broad overview of the field. Examples of specific mAbs are discussed, although only selected references are given because the volume of primary literature on mAbs is immense.
Methods
The results presented here were derived from analysis of a dataset of over 600 therapeutic mAbs that entered clinical study sponsored, at least in part, by commercial firms. Data were collected by survey of pharmaceutical and biotechnology firms and from public documents, e.g., press releases, annual reports and the commercially-available databases IDdb3, IMS R&D Focus and PharmaProjects. Data were updated with all changes noted through July 2010. Data collected included milestone dates, e.g., investigational new drug (IND) application filing, marketing application submission and FDA approval dates; the type of antibody, e.g., murine, chimeric, humanized or human; therapeutic category and FDA review designations, e.g., priority or standard review. The clinical phase was defined as the interval between the earliest of either the IND filing date or the first administration to humans date and the date a marketing application was submitted to the FDA. The clinical phase was thus not limited to development done under a US IND, but was considered complete upon submission to the FDA of the first marketing application for any indication. The therapeutic category was assigned based on the category of the indication first approved.
Entering the Clinic
The rate at which novel antibody therapeutics entered clinical studies per year was relatively flat during the late 1990s and early 2000s (; data presented as a two-year moving average). With the increased participation by major pharmaceutical firms, the rate rose from approximately 20 per year in 2002 to over 50 per year in 2008. The decrease observed in 2009 may be real and possibly a consequence of companies reserving resources during an economic downturn, or it may simply be an undercount because companies, especially major pharmaceutical firms, do not necessarily identify the composition of matter of their Phase 1 candidates.
FDA-Approved mAbs
Clinical phase lengths were derived from data for FDA-approved mAbs. As of July 2010, this cohort included 28 therapeutic mAbs ( and ), but four were voluntarily withdrawn from the US market. The approved products encompass a variety of molecular origins and therapeutic categories. Although the first mAb was approved in 1986, most (93%) were approved after 1996, which is a period during which companies transitioned from the study of murine mAbs to chimeric, humanized and human forms. Of the 28 products, the protein sequences of three (11%) were murine-derived, five (18%) were chimeric, 13 (46%) were humanized and seven (25%) were fully human and derived from either a transgenic mouse or phage display platform.
Clinical phases were calculated for the 23 currently marketed mAbs that were approved after 1996. The mean clinical phase for these products was 85.7 months (). The broad range (37.3–140.3 months) was likely due to the variety of indications studied, which included solid and hematological cancers, disorders of the immune system, infectious disease and eye and bone diseases. Eleven of the 23 products (48%) were first approved as treatments for immunological indications, including psoriasis, asthma, Crohn disease, multiple sclerosis, rheumatoid arthritis (RA), paroxysmal nocturnal hemoglobinuria and prevention of acute kidney rejection. A common feature of these conditions is the need for immune system modulation. The mean clinical phase for the 11 immunological mAbs was 85.6 months and the range was 59.2–113.2 months. Nine of the 23 products (39%) were approved for cancer. These mAbs were first approved as treatments for non-Hodgkin lymphoma, chronic lymphocytic leukemia, breast cancer and colorectal cancer. The mean clinical phase for the nine products was 91.0 months and the range was 50.5–140.3 months.
Only three of the 23 products were approved for indications other than immunological diseases or cancer. Palivizumab (MedImmune) was approved for prevention of respiratory syncytial virus (RSV) infection, ranibizumab (Genentech) was approved for treatment of patients with neovascular age-related macular degeneration and denosumab (Genentech) was approved for postmenopausal osteoporosis. Clinical phases cannot be given for the individual products because the information is proprietary.
Approval phases for the 23 products were affected by whether the candidate received a priority or a standard review by the FDA (). An application is assigned a priority review when the candidate might, if approved, be a safe and effective therapy where none currently exists, or provide significant improvement in disease treatment. If the candidate does not meet either of these criteria, then the application is assigned a standard review. The FDA's current performance goal for priority reviews is 6 months for a first action for 90% of the applications submitted in any given fiscal year. The first action is not necessarily an approval and 10% of the reviews might take longer than 6 months even when the first action is an approval. The majority (70%) of the 23 approved mAbs was priority reviewed; the mean approval phase was 9.3 months () and the range was 4.7–33.4 months for these products. The seven products given a standard review were adalimumab, omalizumab, certolizumab pegol, golimumab, ustekinumab, tocilizumab and denosumab. These products had a mean approval phase of 20.9 months, with a range of 9.1–36.5 months.
Most of the approved mAbs are full-size, human- or murine-derived immunoglobulin gamma (IgG). These are complex molecules composed of multiple protein chains with attached carbohydrates that can be manipulated in various ways to attain desired properties. A full-size IgG antibody is a Y-shaped homodimer of two light chains, each with a variable and constant domain and two heavy chains, each with one variable and three constant domains. The four protein chains have multiple inter- and intra-chain disulfide bridges that maintain the overall conformation of the antibody molecule. Each of the two antigen-binding arms (Fab) of the antibody includes one light chain, as well as the variable and one constant domain of one heavy chain. Through antibody engineering, Fab can be produced instead of the full-size molecule. Of the currently approved products, abciximab, ranibizumab and certolizumab pegol are Fabs.
Four of the 23 approved mAbs were modified to enhance their molecular properties. Three of these are immunoconjugates that include either a toxin (gemtuzumab ozogamicin) or a radiolabel (ibritumomab tiuxetan and tositumomab-I-131). One Fab, certolizumab pegol, is pegylated. Pegylation, i.e., covalent modification of the molecule with polyethylene glycol, alters pharmacokinetic properties such as the circulating half-life of the product.Citation5
Supplemental Approvals
Many mAb products have received at least one supplemental approval from the FDA.Citation6 In some cases the supplemental approvals have simply expanded the basis for treatment of the indication first approved. For example, trastuzumab was first approved in September 1998 as a treatment for metastatic breast cancer in patients whose tumors overexpress HER2 protein and who had received one or more chemotherapy regimens for their metastatic disease. The FDA's approval of trastuzumab as a treatment for breast cancer was expanded twice, once in November 2006 and again in January 2008, to include use of trastuzumab in adjuvant (i.e., post-surgical) combination therapy and as adjuvant monotherapy in early-stage HER2-positive breast cancer. In other cases, supplemental approval has been for a disease in a different therapeutic category compared to that of the indication first approved. For example, rituximab was first approved for treatment of patients with relapsed or refractory low-grade or follicular B-cell non-Hodgkin lymphoma in November 1997, but was given two supplemental approvals for use as a treatment for RA.
The three anti-tumor necrosis factor (TNF) products infliximab, adalimumab and golimumab are notable for the number of indications for which they have been approved by the FDA. Infliximab was first approved in August 1998 as a treatment for moderately-to-severely active Crohn disease, for the reduction of the signs and symptoms in patients who have an inadequate response to conventional therapies and for treatment of patients with fistulizing Crohn disease for reduction in the number of draining enterocutaneous fistulas. A total of 14 supplemental approvals for use of infliximab were subsequently granted: four were for variations of RA treatment (reducing the signs and symptoms of disease, inhibiting structural damage, improving physical function and treatment of early stages not yet treated with methotrexate); two were for psoriatic arthritis (treatment and to improve physical function), one was for active arthritis, including inhibition of structural damage; three were for Crohn disease (pediatric use, reducing signs and symptoms, inducing and maintaining remission and reducing the number of draining fistulas and maintaining fistula closure in fistulating Crohn disease), two were for ulcerative colitis (treatment and maintenance of clinical remission and mucosal healing), one was for ankylosing spondylitis and one was for plaque psoriasis.
Adalimumab, thus far, has fewer supplemental approvals than infliximab. The product was first approved in December 2002 for reducing the signs and symptoms and inhibiting the progression of structural damage in adult patients with moderately-to-severely active RA who have had an inadequate response to one or more disease-modifying anti-rheumatic drugs. Seven supplemental indications were subsequently approved by the FDA: two were for RA (improving physical function in patients and treatment of patients with a recent diagnosis and who are not yet taking methotrexate); one was for juvenile idiopathic arthritis; one was for psoriatic arthritis, including improving physical function and inhibiting structural damage; one was for Crohn disease; one was for ankylosing spondylitis and one was for plaque psoriasis.
Golimumab's biologics license application, which was approved in April 2009, was unusual because it included data for the product as a treatment for three indications: rheumatoid arthritis, psoriatic arthritis and ankylosing spondylitis. The product is currently in Phase 3 studies as a treatment for moderately-to-severely active ulcerative colitis and a Phase 2 study in patients with chronic sarcoidosis. A fourth anti-TNF product, certolizumab pegol, was approved by the FDA in April 2008 for reducing the signs and symptoms of Crohn disease and maintaining clinical response in adult patients with moderately to severely active disease who have had an inadequate response to conventional therapy, but, as of July 2010, the product has not received any supplemental approvals. Certolizumab pegol is currently in Phase 3 studies of patients with rheumatoid arthritis, active axial spondyloarthritis or psoriatic arthritis.
Antibody Products of the Future?
To date, the pharmaceutical industry has focused on clinical development of full size mAbs, but the pipeline now includes in increasing number of antibody fragments.Citation7 As therapeutics, antibody fragments have features that could be advantages or disadvantages compared to those of full-size mAbs. Fragments are smaller and so might potentially penetrate tissues or tumors that could be invulnerable to full-size counterparts. However, fragments degrade faster in humans and have short circulating half-lives, and thus may not accumulate in sufficient quantities at the targeted site.Citation8,Citation9 Modes of action for antibody fragments include blocking the action of biological molecules by binding either ligand or receptor or engaging signaling pathways by cross-linking receptors, but unless designed otherwise, antibody fragments do not induce effector functions such as antibody-dependent cell-mediated cytotoxicity or complement-dependent cytotoxicity.Citation10 Fragments are potentially easier and less costly to manufacture, although they are also more likely to form aggregates and can be less stable than full-size mAbs.
Prior to 1995, development was focused on Fabs, although only one of these was ultimately approved. In 1994, abciximab (Centocor), a Fab that targets glycoprotein IIb/IIIa on platelets, was approved in the US for the prevention of blood clotting in high risk angioplasty. Since then, clinical candidates have included Fabs and a mixture of other engineered formats such as single-chain variable fragments (scFv), although only two Fab have been FDA approved: ranibizumab (Genentech) was approved in 2006 as a treatment for neovascular (wet) age-related macular degeneration (AMD) and certolizumab pegol (UCB) was approved in 2008 for reducing the signs and symptoms of Crohn disease and maintaining clinical response in adults with moderately to severely active disease.
The current pipeline of antibody fragmentsCitation7 includes 24 candidates of which six (25%) are Fab, 13 (54%) are versions of antibody variable fragments and five (21%) are third-generation molecules, which include single domain antibodies composed of one protein chain.Citation11,Citation12 As of July 2010, one single domain, human-derived antibody and three single domain, camelid-derived antibodies were in clinical study. CEP-37247 (Cephalon), an anti-TNF human domain antibody, is currently in Phase 2 studies as a treatment for RA or psoriasis. In addition, three of Ablynx's camelid-derived Nanobodies® are in clinical studies: anti-RANKL ALX-0141 is in Phase 1; anti-von Willebrand factor ALX-0081 is being evaluated in a Phase 2 clinical trial in high risk percutaneous coronary intervention patients and anti-TNF ATN-103 is in Phase 2 studies as a treatment for rheumatoid arthritis. The results of these early stage studies might affect whether third generation fragments will compose a larger percentage of the clinical pipeline in the future.Citation13
Discussion
The field of mAb development is undergoing rapid change. Major pharmaceutical firms, as well as biotechnology companies, are adding substantial numbers of mAbs to their pipelines. Antibody products have established development and approval pathways, with production methods that are well-understood and transferable to contract manufacturers. Research, development and approval phase lengths and approval success rates are competitive with those of small molecule therapeutics.
Comparison of mAbs currently in review with those in review a decade ago illustrates a number of trends. Six mAbs, rituximab, trastuzumab, infliximab, daclizumab, basiliximab and palivizumab, were reviewed by the FDA in the late 1990s. At the time, this group of therapeutics represented important advances in the treatment of serious or life-threatening diseases including lymphoma, breast cancer, Crohn disease, prevention of kidney transplant rejection and prevention of RSV infection. Similar circumstances with regard to the numbers exist today: four mAbs are currently undergoing FDA review for anticancer, immunological and anti-infective indications.
Important distinctions can be made, however, between the late 1990s and the current period. First, whereas the mAbs approved in the late 1990s comprised three chimeric and three humanized products, the current batch includes one humanized and three human products. This fact reflects the current trend toward development of human mAbs, which comprise approximately 45% of the candidates that have entered clinical study in the 2000s.Citation14 Second, a number of the candidates are novel versions of previously approved products: rituximab and ofatumumab are both anti-CD20 mAbs and palivizumab and motavizumab are both anti-RSV mAbs. The existence of these second-generation molecules is a testament to advances in antibody engineering, but also indicates the willingness of companies to directly compete in the current global mAb market.
There are also important differences between the two periods with regard to company pipelines of clinical and preclinical candidates. In the 1990s, the focus was on full-size molecules, whereas there is now a nascent shift toward development of alternative antibody formats and antibody-like scaffolds as therapeutics. Fab and scFv, as well as third generation molecules, are entering clinical study in small but increasing numbers. Additional diversity in molecular types can be seen in the preclinical pipeline.
With the current trend toward study of targeted therapeutics, the focus on human IgG mAbs and alternative antibody formats is likely to intensify due to their desirable characteristics and versatility as therapeutics in a range of indications. The pharmaceutical and biotechnology industry, regulatory agencies, physicians and patients have now gained sufficient experience with mAbs that the treatments are viewed as little different from any other therapeutic. There is as yet significant unmet medical need for new treatments for cancer, immunological and infectious diseases. The innovative antibody therapeutics currently in clinical study for these diseases may bring new hope for patients.
Figures and Tables
Figure 1 Years of first clinical study and approval for therapeutic monoclonal antibodies. *Two-year moving average. FDA, United States Food and Drug Administration.
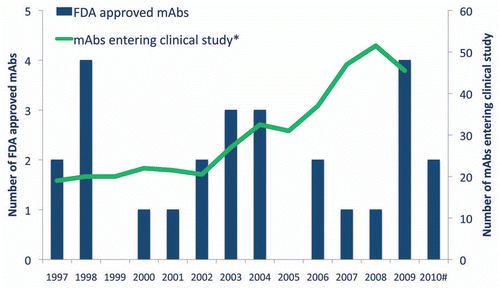
Table 1 Therapeutic monoclonal antibodies undergoing regulatory review or approved in the United States
Table 2 Mean (median) clinical and FDA approval phases for 23 therapeutic monoclonal antibodiesTable Footnote*
Acknowledgements
The author thanks Stella Stergiopoulos for preparation of and colleagues at Tufts Center for the Study of Drug Developement for critical review of the manuscript.
References
- Reichert JM. Monoclonal antibodies as innovative therapeutics. Curr Pharma Biotechnol 2008; 9:423 - 430
- Reichert JM, Rosensweig CJ, Faden LB, Dewitz MC. Monoclonal antibody successes in the clinic. Nat Biotechnol 2005; 23:1073 - 1078
- Reichert JM. Monoclonal antibodies in the clinic. Nat Biotechnol 2001; 19:819 - 822
- Kola I, Landis J. Can the pharmaceutical industry reduce attrition rates?. Nat Rev Drug Discov 2004; 3:711 - 716
- Veronese FM, Mero A. The impact of PEGylation on biological therapies. BioDrugs 2008; 22:315 - 329
- Cohen J, Wilson A. New challenges to medicare beneficiary access to mAbs. mAbs 2009; 1:56 - 66
- Nelson AL, Reichert JM. Development trends for therapeutic antibody fragments. Nat Biotechnol 2009; 27:331 - 337
- King DJ, Turner A, Farnsworth AP, Adair JR, Owens RJ, Pedley RB, et al. Improved tumor targeting with chemically cross-linked recombinant antibody fragments. Cancer Res 1994; 54:6176 - 6185
- Hudson PJ, Souriau C. Engineered antibodies. Nat Med 2003; 9:129 - 134
- Sanz L, Cuesta AM, Compte M, Alvarez-Vallina L. Antibody engineering: facing new challenges in cancer therapy. Acta Pharmacol Sin 2005; 26:641 - 648
- Harmsen MM, De Haard HJ. Properties, production and applications of camelid single-domain antibody fragments. Appl Microbiol Biotechnol 2007; 77:13 - 22
- Saerens D, Ghassabeh GH, Muyldermans S. Single-domain antibodies as building blocks for novel therapeutics. Curr Opin Pharmacol 2008; 8:600 - 608
- Zider A, Drakeman DL. The future of monoclonal antibody technology. mAbs 2010; 2:361 - 364
- Nelson AL, Dhimolea E, Reichert JM. Development trends for human monoclonal antibody therapeutics. Nat Rev Drug Discov 2010; 9:767 - 774