Abstract
The CTLA4-Ig fusion proteins abatacept and belatacept are clinically proven immunosuppressants used for rheumatoid arthritis and renal transplant, respectively. Given that both biologics are typically administered chronically by infusion, a need exists for a next-generation CTLA4-Ig with more convenient dosing. We used structure-based protein engineering to optimize the affinity of existing CTLA4-Ig therapeutics for the ligands CD80 and CD86, and for the neonatal Fc receptor, FcRn. From a rationally designed library, we identified four substitutions that enhanced binding to human CD80 and CD86. Coupled with two IgG1 Fc substitutions that enhanced binding to human FcRn, these changes comprise the novel CTLA4-Ig fusion protein, XPro9523. Compared with abatacept, XPro9523 demonstrated 5.9-fold, 23-fold, and 12-fold increased binding to CD80, CD86, and FcRn, respectively; compared with belatacept, CD80, CD86, and FcRn binding increased 1.5-fold, 7.7-fold, and 11-fold, respectively. XPro9523 and belatacept suppressed human T cell proliferation and IL-2 production more potently than abatacept. XPro9523 also suppressed inflammation in the mouse collagen-induced arthritis model. In cynomolgus monkeys, XPro9523 saturated CD80 and CD86 more effectively than abatacept and belatacept, potently inhibited IgM and IgG immunization responses, and demonstrated longer half-life. Pharmacokinetic modeling of its increased potency and persistence suggests that, in humans, XPro9523 may demonstrate superior efficacy and dosing convenience compared with abatacept and belatacept.
Introduction
Inhibition of costimulatory signals required for T cell activation is now a clinically validated immunosuppressive strategy, with abatacept and belatacept now marketed for rheumatoid arthritis (RA) and kidney transplantation, respectively. Both biologics are fusions of the extracellular domain (ECD) of human CTLA4 (CD152) with the Fc domain of human IgG1. CTLA4, an inhibitory T cell receptor with high homology to the costimulatory receptor CD28, serves as an intrinsic negative regulator of CD28-mediated activation of CD4+ and CD8+ T cells.Citation1 Abatacept and belatacept act by disrupting engagement of CD28 with B7 molecules CD80 and CD86 on antigen-presenting cells (APC).Citation2,Citation3 CTLA4 is also constitutively expressed on regulatory T cells, and consequently several extrinsic immunomodulatory functions that may contribute to the mechanism of action of CTLA4-Ig fusion proteins have been proposed.Citation4,Citation5
A number of biophysical and structural studies have shown that CD86 plays a more significant role than CD80 in engaging CD28 at the T cell—APC synapse,Citation6,Citation7 suggesting that a biologic with increased affinity for CD86 may be a more potent immunosuppressive agent. Indeed, this hypothesis was the rationale for development of belatacept for the prevention of kidney transplant rejection. In contrast to abatacept, which contains a native CTLA4 ECD, belatacept was engineered with two CTLA4 domain substitutions that improve CD80 and CD86 binding by two-fold and four-fold, respectively, resulting in greater suppression of in vitro human T cell responses and of humoral immunity in cynomolgus monkeys.Citation8 In addition, three substitutions were introduced into the Fc domains of both abatacept and belatacept in order to improve protein production and reduce cytotoxic effector functions mediated by engagement of activating Fcγ receptors.Citation9-Citation11
While belatacept is optimized for increased affinity to CD80 and CD86, it shares the same human IgG1 Fc domain with abatacept, and consequently has a similar terminal half-life in humans. It is now well understood that the in vivo half-life of antibodies is regulated by pH-dependent interaction of the Fc domain with the salvage receptor FcRn.Citation12 This discovery has led to efforts to engineer the IgG Fc domain for higher affinity to human FcRn at pH 6.0, with the goal of increasing serum persistence and overall exposure of therapeutic antibodies.Citation13,Citation14 Although this strategy has not yet been applied to Fc fusion proteins, we reasoned that the same Fc substitutions shown to increase persistence of antibodies would also extend the in vivo half-life of CTLA4-Ig. Therefore, in addition to modifying the CTLA4 domain to improve potency, we also engineered the Fc domain to increase affinity for FcRn. The intent of manipulating both domains was to create a CTLA4-Ig biologic with superior efficacy in autoimmune and transplant indications, while offering less frequent dosing to patients on chronic and potentially lifelong treatment regimens.
Results
Engineering CTLA4-Ig for improved affinity to CD80, CD86 and FcRn
The affinity of native CTLA4 and abatacept for CD86 is several fold lower than for CD80,Citation8,Citation15 yet CD86 is thought to be the dominant ligand for CD28-mediated costimulation of T cells.Citation7,Citation16 We thus used a rational structure-based protein engineering approach to improve the binding of CTLA4-Ig to both its ligands, with the emphasis on CD86. Our design efforts were inspired by the previous example of belataceptCitation8 and facilitated by crystal structures for the protein complexes of CTLA4-CD86Citation17 and CTLA4-CD80.Citation18 By examining the interface of CTLA4 with CD80 and CD86, we identified 21 positions as targets for amino acid substitution and used computational modeling to rationally design 131 single-substitution variants of the CTLA4 ECD in an IgG1 Fc fusion format (). These single variants were constructed, expressed, and screened for differences in CD80 and CD86 dissociation rates (koff) relative to abatacept and belatacept (). Of 131 single variants (), the majority of variants preserved or reduced binding to one or both antigens; however, 10 variants had a slower off-rate for CD80, and 13 had a slower off-rate for CD86. Variants with the best improvement in dissociation rate were located at positions A29, T51, M53, L61, K93, and L104 (). Initial hits were subsequently rescreened to determine full KD values for both CD80 and CD86, thereby eliminating seven substitutions from consideration due to a lack of improvement in KD (). The best single substitution variants were confirmed to be A29H, T51N, M53Y, L61E, K93Q, and L104E (one of the substitutions present in belatacept). Notably, several substitutions at position A29 had increased affinity for CD86, including H, K, W, and Y, with A29H having tighter and more selective binding to CD86 than the A29Y substitution found in belatacept. Selectivity was greatest with the T51N substitution, which showed a 2.6-fold improvement in CD86 binding, with a concomitant 2.3-fold reduction in CD80 binding. We combined the five substitutions with tighter binding to CD86 to generate 18 variants with 2–5 substitutions each, and then determined KD values in another round of screening (). Of these 18 variants, all had higher affinity to CD86 compared with abatacept, and the combination of A29H, T51N, L61E, and K93Q (abbreviated HNEQ) generated the best target binding profile, with 23-fold and 5.9-fold improved affinity to CD86 and CD80, respectively, compared with abatacept. HNEQ also showed 7.7-fold higher affinity to CD86 and 1.5-fold higher affinity to CD80 compared with belatacept.
Figure 1. Engineering CTLA4-Ig variants for increased binding to CD80 and CD86. (A) and (B) Using known complex structures, Citation17,Citation18 CTLA4 molecules (blue) are shown interacting with CD80 (A), dark magenta) and CD86 (B), light magenta). CTLA4 residues within contact distance of CD86 targeted for substitution are shown in stick format, along with neighboring residues in CD80 or CD86. Six positions at which substitutions significantly improved affinity for CD80 or CD86 are labeled. (C) Dissociation rates (koff) of CTLA4-Ig variants for CD80 and CD86, relative to abatacept. Shown are abatacept (open square, obscured on the 1-fold CD80-CD86 intercept), belatacept (open circle), and 131 single substitution CTLA4-Ig variants (solid diamonds), including the five listed variants with the highest increase in affinity. (D) Equilibrium dissociation constants (KD) of CTLA4-Ig variants for CD80 and CD86, relative to abatacept. Shown are abatacept (open square at intercept), belatacept (open circle) and 17 multiple-substitution CTLA4-Ig variants (solid diamonds). HNEQ (star) is the selected precursor CTLA4-Ig containing the four substitutions (A29H, T51N, L61E, K93Q) present in XPro9523. (E) Summary of binding data for best single and combination CTLA4-Ig variants. Fold improvements in KD relative to abatacept are shown for CD80 (black bars) and CD86 (gray bars). Bars are overlapping, and belatacept is included for comparison.
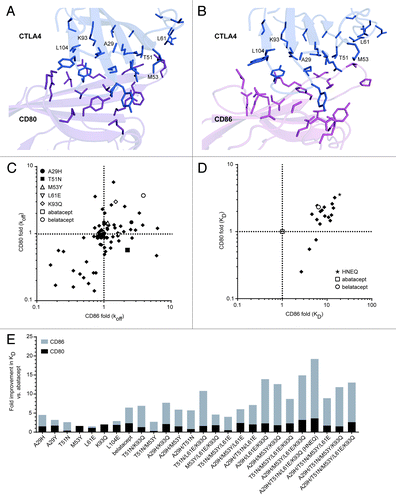
Table 1. Summary of changes in dissociation rate for CTLA4-Ig variants binding to CD80 or CD86
To improve the pharmacokinetic properties of CTLA4-Ig, we also optimized the affinity at pH 6.0 of its IgG1 Fc domain for the human neonatal receptor FcRn. We have previously shown that, in the context of full-length antibodies, the Fc substitutions M428L and N434S improve affinity to FcRn by 11-fold, resulting in increased serum half-life in cynomolgus monkeys.Citation13 We reasoned that this modification would also improve the half-life of an Fc fusion protein, so we introduced M428L and N434S substitutions into the Fc region of HNEQ to generate XPro9523. This resulted in an approximately 11-fold improvement in FcRn binding relative to abatacept and belatacept while maintaining the improved binding to CD80 and CD86 introduced by the engineered CTLA4 ECD ( and ). Notably, XPro9523 also contains the C220S, C226S, and C229S substitutions introduced into the IgG1 Fc domain of both abataceptCitation9 and belatacept.Citation10 As reported for CTLA4-Ig fusions,Citation9 these substitutions are known to minimize potential cytotoxicity mediated by engagement of FcγRIIa and FcγRIIIa activating receptors on immune effector cells.
Figure 2. Surface plasmon resonance (SPR) analysis of CTLA4-Ig binding to CD80, CD86, and FcRn. SPR traces are shown for abatacept, belatacept, and XPro9523 binding to CD80 (top), CD86 (middle), and FcRn (bottom). SPR binding curves for CD80 were performed at 0.313, 0.625, 1.25, 2.5, 5.0, and 10.0 nM; binding curves for CD86 were performed at 0.078, 0.156, 0.313, 0.625, 1.25, and 2.5 nM; and binding curves for FcRn were performed at 62.5, 125, 250, and 500 nM.
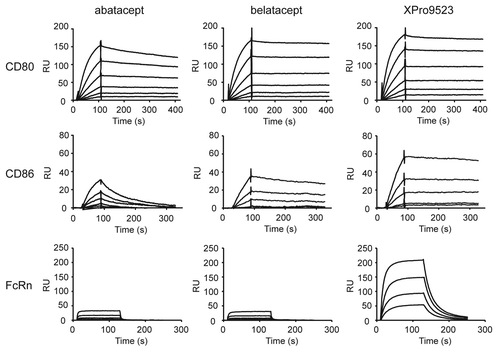
Table 2. Equilibrium dissociation values for abatacept, belatacept, and XPro9523 binding to CD80, CD86, and FcRn
XPro9523 inhibits T cell proliferation and IL-2 induction in vitro
Based on previous CTLA4-Ig engineering efforts,Citation8 the increased affinity of XPro9523 for CD86 is predicted to more potently suppress T cell costimulation compared with abatacept. We therefore tested the ability of XPro9523, abatacept, and belatacept to inhibit human T cell proliferation induced by recombinant CD86. T cells isolated from human peripheral blood mononuclear cells (PBMCs) were stimulated with anti-CD3 and anti-CD86 in the presence of increasing concentrations of CTLA4-Ig biologics. XPro9523 was 20-fold more potent at inhibiting CD86-mediated T cell proliferation compared with abatacept, and two-fold more potent than belatacept (). The relative potencies of T cell proliferation induced by CD86 cross-linking are broadly consistent with the relative affinities of these three CTLA4-Ig biologics for CD86.
Figure 3. Activity of XPro9523 relative to abatacept and belatacept in in vitro human T cell proliferation and mixed lymphocyte reaction (MLR) assays. (A) T cell proliferation. T cells were isolated from PBMCs and stimulated with anti-CD3 and recombinant CD86 in the presence of CTLA4-Ig, and incubated for 4 d prior to quantification of T cell number. The percent proliferation was determined as the fraction of cells with a decrease in CFSE mean fluorescence intensity. (B) MLR. PBMCs from two different donors were mixed in the presence of CTLA4-Igs, and IL2 induction was measured after 6 d. Values in (A) and (B) represent mean ± SEM of 2 replicates. Data shown in (A) are representative of two independent experiments, and in (B) are representative of at least 6 independent studies.
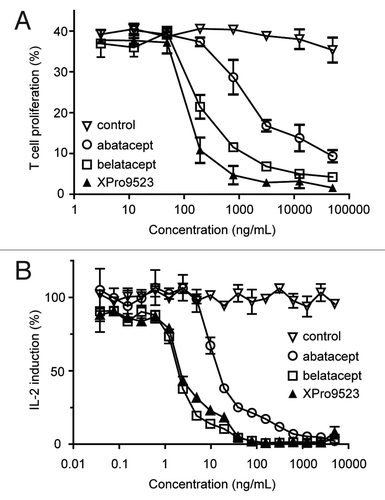
We also compared the ability of XPro9523, abatacept, and belatacept to inhibit production of IL-2 in a mixed lymphocyte reaction (MLR) assay. T cells were stimulated by co-culture of PBMCs from two different donors, with allogeneic APC providing CD80 and CD86 costimulatory signals. Measurement of IL-2 levels as a marker of T cell activation showed that XPro9523 was ~5-fold more potent than abatacept at inhibiting primary allostimulation in vitro (). However, in contrast to the direct T cell proliferation assay, XPro9523 and belatacept showed similar potency, which highlights differences in the complexity of these assays. Whereas the T cell proliferation assay uses a fixed concentration of recombinant CD86 to stimulate T cells with no CD80 present, the MLR assay requires interaction between T cells and multiple types of APC, all with dynamic CD80 and CD86 expression levels, in which the relative importance of CTLA4-Ig affinity for each B7 molecule remains unknown.
XPro9523 suppresses collagen-induced arthritis in mice
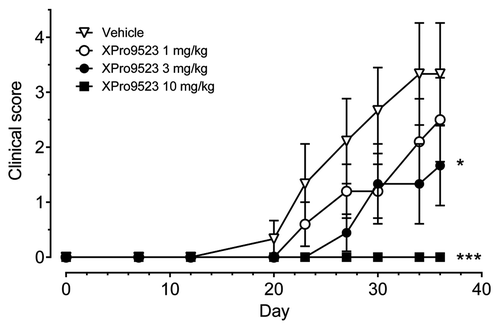
Human CTLA4-Ig binds to murine CD80 and CD86 and has been proven efficacious in multiple animal models of inflammation, including collagen-induced arthritis;Citation19-Citation21 however, optimization of CTLA4-Ig for enhanced potency in humans does not correlate well in mice. For example, belatacept has 2-fold and 15-fold lower affinity for mouse CD80 and CD86, respectively, relative to abatacept (Fig. S1A and B). HNEQ, with even higher affinity for human CD80 and CD86 than belatacept, shows 11-fold reduced binding to mouse CD80, but 1.7-fold increased binding to mouse CD86 (Fig. S1A and B). Surface binding to CD80 and CD86 on mouse B cells (Fig. S1C and D) correlates well with the differences in receptor binding affinity observed for the three CTLA4-Igs. Likewise, enhancement for human FcRn does not translate into longer half-life in mice. Native human IgG1 Fc has 9-fold higher affinity for mouse FcRn than for human FcRn;Citation22 unfortunately, the 11-fold higher affinity for human FcRn enabled by the M428L and N434S substitutions in human IgG1 decreases half-life in mice (data not shown). The disparate affinities of XPro9523, abatacept and belatacept for mouse CD80, CD86 and FcRn therefore preclude meaningful comparison of these human biologics in murine disease models. Given the common use of murine systems to evaluate therapies for human disease, however, we assessed XPro9523 efficacy in the mouse collagen-induced arthritis (CIA) model. XPro9523 at 1, 3, and 10 mg/kg produced a dose-dependent and significant effect on clinical score in DBA/1 mice, with the 10 mg/kg dose completely suppressing inflammation (). This demonstrates that, regardless of its affinity optimization for three human receptors, XPro9523 can be used to study B7 inhibition in traditional mouse models of inflammation and autoimmunity.
Figure 4. Suppression of mouse collagen-induced arthritis by XPro9523. XPro9523 was dosed twice weekly at 1, 3, or 10 mg/kg starting 2 d before immunization with collagen. Clinical score represents sum of swelling of four paws on a scale of 0 (normal) to 3 (severe). Values represent mean ± SEM from 10 mice per group. The p values shown represent 1-way repeat-measures ANOVA with Dunnett's post-test vs. PBS-treated group (*p < 0.05; ***p < 0.001).
XPro9523 has higher receptor occupancy and inhibits primary and secondary immune responses more potently than abatacept in cynomolgus monkeys
The goal in engineering a CTLA4-Ig variant with higher affinity to CD80, CD86, and FcRn was to create a more immunosuppressive biologic that also offered the potential for an extended dosing interval. In cynomolgus monkeys, affinity for FcRn is also increased 11-fold, resulting in improved half-life of M428L and N434S-substituted antibodies.Citation13 Because cynomolgus monkeys have been established as a relevant species to investigate immune suppression by both abatacept and belatacept,Citation8,Citation23 we dosed monkeys with 6 mg/kg of either XPro9523 or abatacept and then immunized animals with KLH. Receptor occupancy of CD80 and CD86, as well as anti-KLH IgG and IgM and drug levels in serum, were monitored throughout the study. XPro9523 showed a rapid and sustained saturation of both CD80 and CD86 receptors at all time points examined; in contrast, receptor occupancy by abatacept decreased over time and had returned nearly to control levels after 16 d (). Although XPro9523 target coverage was more effective than abatacept at all time points, the difference was greatest at 23 d, suggesting that the two Fc substitutions of XPro9523, coupled with its higher affinity for CD80 and CD86, might prolong suppression of immune responses.
Figure 5. Receptor occupancy and inhibition of immune responses to KLH in monkeys given PBS, abatacept or XPro9523 (6 mg/kg). (A) CD80 (top) and CD86 (bottom) occupancy on CD20+ B cells measured in whole blood following administration of vehicle or CTLA4-Ig biologics. CD20+ B cells were identified in the lymphocyte gate as determined from forward vs. side scatter plots. Receptor occupancy is expressed as the percent of unbound CD80 or CD86 relative to pre-treatment baseline. (B) Suppression of primary and secondary antibody responses to KLH administered 24 h and 16 d after administration of vehicle or CTLA4-Ig biologics. Top graphs, anti-KLH IgG levels and bottom graphs, anti-KLH IgM levels at 11 d, 21 d, and 27 d following administration of vehicle or CTLA4-Ig. In (A) and (B) values represent mean ± SEM from 4 monkeys. The p values shown represent one-way ANOVA with Bonferroni’s post-test; *p < 0.05; **p < 0.01; ***p < 0.001; if unlabeled, not significant (p > 0.05). (C) Pharmacokinetic data. Left, serum concentrations over time. Right, half-life, area under the curve (AUC), and clearance (mean ± SEM of 4 monkeys). The p values shown represent unpaired t tests.
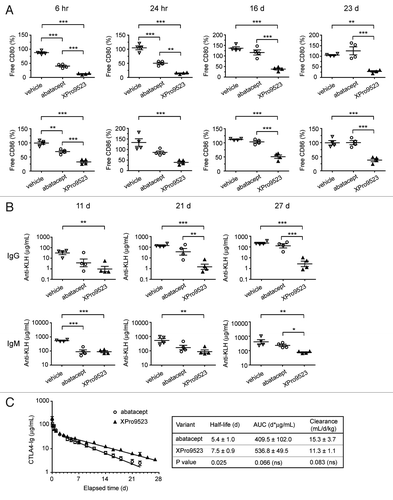
To evaluate the relationship between CD80 and CD86 receptor occupancy and suppression of primary and secondary immune responses, we administered KLH 24 h after the single dose of CTLA4-Ig biologics or vehicle, and boosted again 15 d later. The amount of anti-KLH IgG and IgM present at the peak of the primary response (11 d), the peak of the secondary response (21 d), and at the end of the study (27 d) is shown in . XPro9523 suppressed the IgG immune response to KLH by 33, 95, and 85-fold, and the IgM immune response by 6, 6, and 5-fold relative to vehicle at 11 d, 21 d, and 27 d, respectively. XPro9523 was significantly more effective than abatacept in suppressing IgG responses at 21 d (25-fold) and 27 d (47-fold), and showed greater suppression even at 11 d (4-fold). Results for IgM were similar but less pronounced (3-fold) at 27 d, with no difference seen at 11 d. Sustained suppression of both IgG and IgM anti-KLH antibody levels at later time points correlated well with the higher and extended receptor occupancy observed for XPro9523.
In the same study, we also determined serum levels of XPro9523 and abatacept. Both biologics showed a typical two-phase clearance. The half-life of XPro9523 was 39% longer than abatacept and exposure was 31% greater due to a 26% reduction in clearance (), demonstrating that the serum half-life of an Fc-fusion protein can be extended by increasing its affinity to FcRn.
Comparison of receptor occupancy and immune responses after a single dose of XPro9523 or belatacept in cynomolgus monkeys
Given the prolonged efficacy of high-dose XPro9523 relative to abatacept, in a second monkey study, we compared XPro9523 with the second-generation CTLA4-Ig, belatacept. We intentionally designed this study to assess suboptimal receptor occupancy, comparing pharmacodynamics of a single low dose (0.1, 0.5 or 2.0 mg/kg) of XPro9523 and belatacept (for comparison, abatacept and belatacept are dosed in humans at approximately 10 mg/kg). XPro9523 and belatacept occupied CD80 and CD86 receptors in a dose-dependent manner at all time points (). Both biologics showed similar initial target coverage of CD80 and CD86, though XPro9523 caused greater CD86 saturation at the highest dose, particularly at the 1, 16, and 23 d time points.
Figure 6. CD80 and CD86 receptor occupancy measured in whole blood from monkeys given PBS, belatacept, or XPro9523 (0.1, 0.5, or 2.0 mg/kg). Blood was stained with anti-human CD80 and CD86 antibodies cross-reactive with monkey antigens to compete with CTLA4 binding, as determined by anti-CD80 and anti-CD86 mean fluorescence intensity of CD20+ B cells after standardization for the number of antibodies bound. CD20+ B cells were identified in the lymphocyte gate as determined from forward vs. side scatter plots. Receptor occupancy is expressed as the percent of unbound CD80 or CD86 relative to pre-treatment baseline. Shown are percent free CD80 (left panels) and CD86 (right panels) on B cells at 6 h, 24 h, 16 d, or 23 d following administration of vehicle or CTLA4-Ig. Values represent mean ± SEM from 4 monkeys. The p values shown are for vehicle vs. the indicated dose of CTLA4-Ig, or for XPro9523 vs. belatacept (shown by bracket), and represent one-way ANOVA with Bonferroni’s post-test. *p < 0.05; **p < 0.01; ***p < 0.001; if unlabeled, not significant (p > 0.05).
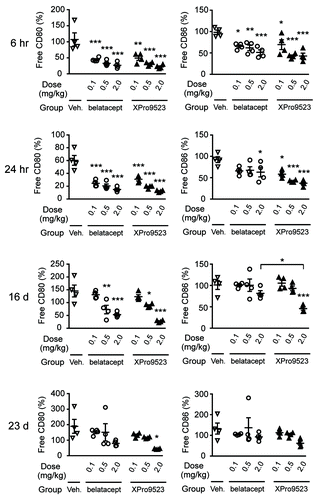
To assess if receptor occupancy correlated with suppression of primary and secondary immune responses, we immunized monkeys with KLH 24 h after treatment with XPro9523 or belatacept. Inhibition of anti-KLH IgG and IgM was evident during the primary response at 11 d, and the single dose of biologic continued to inhibit after the KLH boost given at 16 d (). For both molecules, there was a clear dose response for inhibition of the IgG secondary response at 21 d. Suppression of the IgM response was also evident, but in agreement with the previous study, more modest for both XPro9523 and belatacept.
Figure 7. (A) Suppression of primary and secondary antibody responses to KLH in monkeys given PBS, belatacept, or XPro9523 (0.1, 0.5, or 2.0 mg/kg). KLH was administered 24 h and 16 d after administration of vehicle or CTLA4-Ig, and serum anti-KLH was determined using ELISA. Shown are anti-KLH IgG and IgM levels at 11 d (top panels) and 21 d (bottom panels) following administration of vehicle or CTLA4-Ig. Values represent mean ± SEM from 4 monkeys. The p values shown are for vehicle vs. the indicated dose of CTLA4-Ig and represent one-way ANOVA with Bonferroni’s post-test. *p < 0.05; **p < 0.01; ***p < 0.001; if unlabeled, not significant (p > 0.05). There was no statistical significance between belatacept and XPro9523 at any dose. (B) Pharmacokinetic data for 0.1, 0.5, or 2.0 mg/kg belatacept or XPro9523 administered to monkeys as a 1 h infusion. Shown are serum concentrations over time of abatacept and XPro9523 (values represent mean ± SEM of 4 animals). Pharmacokinetic parameters were determined for individual monkeys with a non-compartmental model. Half-lives were dose-dependent, with means of 3.7 ± 1.1 and 5.1 ± 0.3 d for belatacept and XPro9523 at the 2 mg/kg dose, respectively, suggesting a target sink effect.
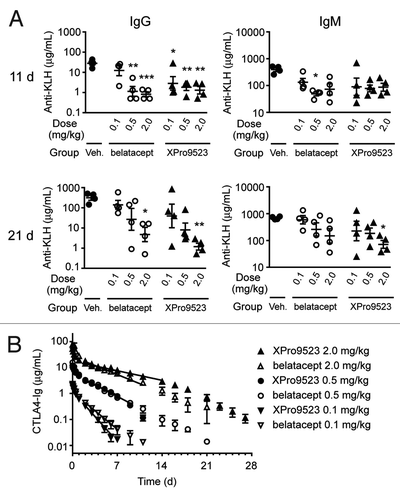
Serum levels of XPro9523 and belatacept were similar at the three doses tested (). It is noteworthy that XPro9523 showed a slightly shorter serum half-life than belatacept at 0.1 mg/kg, a similar half-life at 0.5 mg/kg, and a slightly longer half-life at 2.0 mg/kg. When taken together with the extended half-life for XPro9523 at 6.0 mg/kg shown in , this suggests that higher affinity for CD80 and CD86 increases the antigen sink for CTLA4-Ig biologics, with effects on serum levels most noticeable at doses below target saturation (as shown in ). At the lowest concentration of 0.1 mg/kg (100-fold lower than clinical dosing), this manifests as a shorter apparent half-life for XPro9523. At 0.5 mg/kg, this sink is balanced by enhanced FcRn-mediated recycling, resulting in equivalent apparent half-life; while at the highest concentrations of 2 mg/kg and 6 mg/kg, the half-life of XPro9523 is longer than belatacept () and abatacept (), respectively. In support of the target sink hypothesis, at the 2.0 mg/kg dose in , the half-life and volume of distribution for XPro9523 and belatacept were 5.1 d and 87.1 ml/kg, and 3.8 d and 57.2 ml/kg, respectively. This suggests that in the current clinical regimen of 10 mg/kg dosing for both approved CTLA4-Ig biologics, the observed differences in half-life and volume of distribution caused by FcRn engineering may be further accentuated.
Simulation of human dosing of XPro9523 and comparison to abatacept and belatacept
The 11-fold higher affinity for FcRn at pH 6.0 and the resulting increase in half-life observed in monkeys suggests that XPro9523 could be dosed less frequently yet still maintain effective drug exposure and CD80 and CD86 receptor occupancy. To explore various dosing regimens, we developed a set of two-compartment models describing the human dosing of XPro9523, abatacept, and belatacept. Half-lives and volumes of distribution for abatacept (13.1 d, 70 ml/kg) and belatacept (9.8 d, 110 ml/kg) were drawn from the full prescribing information for Orencia®Citation24 and Nulojix®,Citation25 respectively; other necessary pharmacokinetic parameters were estimated. Model parameters describing XPro9523 were assumed to be equal to those describing abatacept with the key exception of a 39% longer half-life (18.2 vs. 13.1 d), an assumption based on the results of the monkey studies above.
First, we explored whether XPro9523 could be dosed more conveniently than abatacept in the RA setting. The approved intravenous (i.v.) dosing regimen for abatacept in RA is an approximate 10 mg/kg dose given every 2 wks for the first 4 wks and every 4 wks thereafter; this leads to serum levels well above the desired 10 μg/ml trough or minimum effective concentration (MEC).Citation26 Our initial approach was to match the serum concentration and drug exposure seen for abatacept, assuming that the 6- and 20-fold higher affinity for CD80 and CD86, respectively, would lead to greater immunosuppression for a given exposure level. Simulations revealed that XPro9523 could be dosed less frequently (every 6 wks vs. every 4 wks), without the need for a loading phase, while still maintaining serum levels above 10 μg/ml (). Another approach we explored was to maintain XPro9523 serum levels above a MEC of 1 μg/ml, as experimental results described above indicate that XPro9523 may offer at least 10-fold more potent suppression than abatacept. Further simulations revealed that a 10 mg/kg i.v. dose would need to be administered only every 3 mo to maintain XPro9523 trough serum concentrations at least 4-fold above this predicted MEC (). Next, because abatacept has recently been approved for weekly 125 mg subcutaneous (s.c.) dosing in RA,Citation24 we modeled less frequent s.c. dosing regimens for XPro9523. The results suggested that XPro9523 could be dosed s.c. every 2 wks or every 4 wks at 125 mg (modeled as a 1.67 mg/kg dose, assuming a 75 kg patient) while maintaining serum concentrations above 10 μg/ml or 1 μg/ml, respectively ().
Figure 8. Simulated human pharmacokinetics of XPro9523. (A) I.V. dosing in RA. Abatacept is given at 10 mg/kg every other week for 4 wks, and every 4 wks thereafter (red line), generating serum levels well above the desired 10 μg/ml MEC (dashed black line). Simulations show that XPro9523 (black line) could be dosed every 6 wks while maintaining exposure comparable to abatacept. In simulations including the additional potency observed for XPro9523, dosing every 3 mo (green line) maintains trough concentrations ~4-fold above the anticipated MEC of 1 μg/ml (dashed green line). (B) S.C. dosing in RA. Simulations indicate that XPro9523 could be dosed every 2 wks (black line) or 4 wks (green line) (modeled as 125 mg in a 75 kg patient) while maintaining serum levels greater than the 10 or 1 μg/ml predicted MEC, respectively (dashed black and green lines). (C) I.V. dosing in renal transplant. Belatacept is administered as 10 mg/kg doses on days 1 and 5 and weeks 2, 4, 8, and 12, followed by maintenance dosing of 5 mg/kg every 4 wks. Simulations revealed that XPro9523 (black line) could be dosed much less frequently while maintaining exposure comparable to belatacept (orange line) and well above the desired MEC of 20, 5, and 2 μg/ml during months 1, 2 to 3, and 4 to 6, respectively, after transplantation (dashed black line). Notably, doses at 2, 8, and 12 wks would be unnecessary for XPro9523, and maintenance dosing could be every 12 instead of every 4 wks
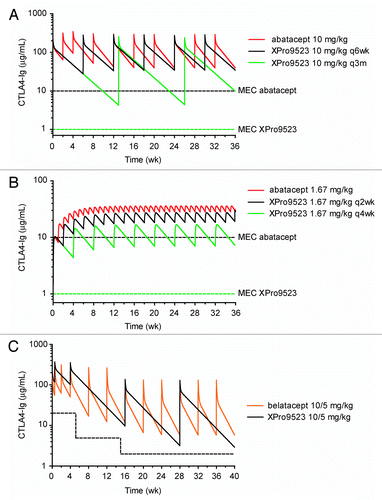
Finally, we examined whether the projected 18.2-d half-life of XPro9523 vs. the 9.8-d half-life of belatacept would allow for more convenient dosing for kidney transplant recipients. The approved i.v. dosing regimen for belatacept in transplantation is a series of 10 mg/kg doses (days 1 and 5, weeks 2, 4, 8 and 12) followed by a maintenance phase of 5 mg/kg doses starting at week 16 and every 4 wks thereafter. This regimen is intended to meet or exceed trough concentrations of 20, 5, and 2 μg/ml during month 1, months 2 to 3, and months 4 to 6 after transplantation, respectively.Citation27 Taking into account its longer half-life, but discounting its increased target affinity relative to belatacept, simulations revealed that XPro9523 could be dosed much more infrequently while maintaining exposure and serum levels comparable to belatacept (). Strikingly, it was projected that doses at weeks 2, 8, and 12 would not be necessary, and the 5 mg/kg maintenance phase could be dosed every 12 wks instead of every 4 wks.
Discussion
With abatacept and belatacept approved for RA and renal transplant rejection, respectively, CTLA4-Ig is a prominent example of the potential of protein engineering to create superior therapeutics. Abatacept, the first such biologic to be developed, incorporates an unmodified human CTLA4 domain and therefore has native binding affinity for CD80 and CD86. The developers of abatacept observed that it was relatively ineffective in nonhuman primate transplant modelsCitation23,Citation28 and showed that the increased potency engineered into belatacept markedly improved efficacy in these challenging models.Citation8 Interestingly, the only published report comparing therapeutic efficacy of abatacept and belatacept describes an RA trial involving 214 patientsCitation29 in which both biologics demonstrated efficacy vs. placebo. The study was not powered to reveal differences between the biologics, and the authors noted that abatacept, rather than the second-generation molecule belatacept, was selected for further development in RA, without elaborating on the basis for this choice. However, the subsequent development and approval of the more potent molecule for renal transplant validates a key assumption in our engineering efforts, which is that increasing the affinity of a CTLA4-Ig biologic for CD80 and CD86 enhances immunosuppression.
We used available structures of CTLA4 complexed with CD80 and CD86 to rationally engineer CTLA4-Ig to improve binding affinity beyond that of abatacept and belatacept. Encouragingly, our screening corroborated the key substitutions A29Y and L104E originally identified during generation of belatacept,Citation8 but our approach identified A29H not only as superior to A29Y, but also as the best single variant, with good selectivity for CD86 relative to CD80. This may be explained by the central location of A29 in the interface of CTLA4 with CD80 and CD86. The β carbon of A29 is pointed up and away from CD80 (), while pointed down toward CD86 (). Substitution with histidine at this position may create a hydrogen bond with the side chain of a neighboring serine present in CD86, but not in CD80. Substitution with the larger residue may also cause a reorientation of the CDR3-like loop of CTLA4, pushing it closer to CD86 and thus leading to a tighter interaction. Conversely, the loop region 25SPGKCitation28 is disordered in the CD86 complex, and substitution with a larger residue may anchor this loop in a more ordered conformation, allowing more interactions with CD86. T51N may also make several new interactions with the main chain of an extended loop in CD86. Of note, L61 was identified during our structural analysis as being the most likely target to improve CD86 binding; therefore, we chose to make all possible amino acid substitutions (with the exception of cysteine) at this position. Most substitutions were neutral, but the side chains of aspartic acid and especially glutamic acid were superior, most likely due to hydrogen bonding with an adjacent threonine side chain on CD86. Finally, the K93Q substitution may be interacting with the same arginine on CD86 that interacts with the L104E substitution because 104E most likely results in formation of a new salt bridge, while 93Q most likely reduces repulsion between the lysine and arginine side chains. Interestingly, the combination of the five best single substitutions (HNEQ plus M53Y) did not generate the highest affinity variant. This result, and our finding that M53Y/T51N showed the smallest improvement in KD of all double variants examined, suggests that the larger side chains of the M53Y and T51N substitutions negatively interact.
It is well-established that improving Fc affinity for FcRn at pH 6.0 results in longer serum half-life and greater overall exposure of antibody therapeutics.Citation14 Notably, this has been demonstrated for the specific Fc domain incorporated into XPro9523.Citation13 Although FcRn optimization has not previously been applied to Fc fusion proteins such as belatacept and abatacept, it was logical to simultaneously optimize both Fc and CTLA4 domains to create a third-generation biologic with the prospect of superior efficacy coupled with improved dosing convenience. Because we did not assess the effects of Fc substitutions independently of CTLA4 optimization (e.g., in the context of abatacept or belatacept), we cannot definitively ascribe increases of in vivo half-life and efficacy solely to higher affinity for FcRn and CD80-CD86, respectively. However, the in vitro human T cell activation results (in which FcRn presumably plays no role), coupled with the longer half-life and extended immunosuppression observed in monkeys, suggests that both FcRn and CD80-CD86 interactions contribute to the improved efficacy of XPro9523 relative to abatacept and belatacept. Modeling of human dosing based on our results suggests that relative to both belatacept and abatacept, XPro9523 may be dosed 3- to 4-fold less frequently while maintaining or increasing drug exposure, target receptor occupancy and predicted efficacy.
Our investigation of the binding of optimized human CTLA4-Ig to mouse CD80 and CD86 illustrates one problem with assessing human therapeutic proteins in murine models. Human CTLA4-Ig biologics, including abatacept, are efficacious in murine disease models such as CIA and NZB/NZW lupus,Citation19-Citation21 and our results for XPro9523 in CIA () corroborate these reports. However, the noncorrelative data for abatacept, belatacept, and HNEQ binding to human and mouse CD80 and CD86 (Fig. S1) demonstrate that optimization for human targets does not translate well to the mouse. Our results suggest that given adequate target cross-reactivity, mice may be suitable for mechanism-of-action studies, but are unlikely to be informative in comparisons of biologics affinity-optimized for human targets. Although transgenically humanized mice are sometimes offered as a solution to this problem, CTLA4-Ig biologics represent a particularly challenging case; such mice would need to incorporate not only functional human CD80 and CD86 receptors, but also FcRn and any other human proteins suspected to interact with CTLA4 and IgG1 Fc.
All biologics have the potential for immunogenicity, and this is a particular concern for chronic dosing of non-native proteins such as Fc fusions. Abatacept contains four substitutions relative to an Fc fusion possessing native CTLA4 and IgG1 Fc domains, and belatacept and XPro9523 are engineered with two and five additional amino acid changes, respectively, relative to abatacept. Because the potency of T cell inhibition is substantially increased for belatacept and XPro9523, they likewise have greater potential to suppress immune responses to their own administration. In support of this possibility, only 2% of patients in clinical trials developed antibodies against belatacept, which was comparable to the 1.7% reported for abatacept.Citation24,Citation25 Thus, the additional mutational load of XPro9523 is not predicted to markedly alter immunogenicity, although this ultimately must be assessed in the clinic. Other potential side effects are expected to be related to the immunosuppressive mechanism of action of CTLA4-Igs such as abatacept and belatacept, which are known to increase the risk of infections.Citation24,Citation25
In summary, targeting T cell costimulatory receptors with CTLA4-Ig biologics is now established as an effective immunosuppression strategy. The combination of higher potency plus longer half-life engineered into XPro9523 as a third-generation CTLA4-Ig may facilitate new treatment regimens not only in RA and transplant, but also in other autoimmune diseases where the existing therapeutic options are inconvenient or ineffective.
Materials and Methods
Protein engineering
DNA encoding the CTLA4 ECD of abataceptCitation9 and belataceptCitation8 was generated by gene synthesis (Blue Heron Biotechnologies) and cloned into the pTT5 expression vector (National Research Council Canada) encoding the human IgG1 Fc region heavy chain. All clones included the C220S, C226S, and C229S Fc substitutions that were previously incorporated into both abatacept and belatacept to minimize affinity for Fcγ receptors.Citation9,Citation10 The proline residue at position 238, originally introduced as a cloning artifact and still present in abatacept and belatacept,Citation10,Citation11 was corrected to the native serine residue in our CTLA4 library. Biologics were produced in HEK 293E cells for initial screening and CHO-3E7 cells for in vivo studies (both cell lines were obtained from National Research Council Canada), purified by protein A chromatography, and analyzed by SDS-PAGE and SEC for correct size and homogeneity. Using rational structure-based engineering, 21 positions in the ECD of CTLA4 were identified as targets for mutagenesis. A total of 149 variants were constructed at these positions during two rounds of affinity optimization. Substitutions were introduced into the CTLA4 ECD of abatacept using site-directed mutagenesis (QuikChange, Stratagene) and variants were expressed, purified, and characterized as described above. CTLA4-Ig variants were analyzed for CD80 and CD86 antigen binding by surface plasmon resonance (SPR) measurement with a BIAcore 3000 instrument (GE Healthcare). Polyhistidine-tagged CD86-Fc and CD80-Fc were purchased from R&D Systems and immobilized on an anti-His CM5 chip at 100 nM for 1.5 min, followed by injection of 2-fold serial dilutions of CTLA4-Ig (0.3–10 nM for CD80; 0.8–2.5 nM for CD86) for 1.5 min, with a 6 min dissociation phase. Equilibrium dissociation constants were calculated using a simple 1:1 Langmuir binding model. Substitutions M428L and N434S were introduced into the IgG1 Fc domain of CTLA4-Ig variants to improve binding to FcRn, and affinity was quantified as described.Citation13
In vitro T cell proliferation and mixed lymphocyte reaction assays
For T cell proliferation assays, 96-well plates were coated with 2 µg/ml CD86 and 0.5 mg/ml anti-CD3 in PBS at 4°C overnight, then washed 3 times with PBS. Human T cells purified from PBMCs using a negative enrichment kit (STEMCELL Technologies) were labeled with 10 µM carboxyfluorescein succinimidyl ester (CFSE) and 5 × 105 cells were added to each well. CTLA4-Ig variants or control (anti-respiratory syncytial virus IgG1) were added starting at 50 µg/ml with 8 subsequent 4-fold dilutions in duplicate, and plates were incubated at 37°C. After 4 d, CFSE fluorescence was used to quantify cell proliferation using a FACSCanto II flow cytometer (BD Biosciences). For MLR assays, 3 × 105 PBMCs from two different donors were mixed in 200 µL RPMI1640 containing 10% FBS. Varying concentrations of CTLA4-Ig variants were added, cells were incubated at 37°C for 6 d, and IL-2 production was quantified using an IL-2 ELISA MAX Kit (Biolegend).
Collagen-induced arthritis in mice
Male DBA/1 mice (Taconic), 11 wks old, were injected s.c. at two sites (base of tail and back) with 0.2 mg chicken collagen in CFA and boosted on day 14, using IACUC-approved protocols essentially as described.Citation30 Mice were injected intraperitoneally with 1, 3, or 10 mg/kg XPro9523 twice per week starting 2 d before collagen immunization. Paw swelling was measured using digital calipers (MSI Viking Gage), and clinical scores were determined in a blinded fashion as the sum of 4 paws ranging from 0 (normal paw) to mild (1), moderate (2) and severe (3) swelling.
Comparison of XPro9523 and abatacept in cynomolgus monkeys immunized with KLH
Monkey studies were conducted at SNBL USA using IACUC-approved protocols. All test articles were well-tolerated, and animals were returned to colony stock upon study completion. Female cynomolgus monkeys (Macaca fascicularis) weighing 2.7–3.5 kg were randomized by weight, divided into 3 groups (n = 4) and given a single, 1 h i.v. infusion of PBS or 6 mg/kg of XPro9523 or abatacept in a dose volume of 5 ml/kg. Keyhole limpet hemocyanin (KLH) (3 mg, Sigma-Aldrich) was administered intramuscularly 24 h after the end of infusion, and a 3 mg boost was given after 16 d. Blood samples (1 ml) were drawn and processed to serum for pharmacokinetic and pharmacodynamic analyses. For determination of CD80 and CD86 receptor occupancy attributable to CTLA4-Ig binding of monkey B cells, whole blood samples in sodium citrate were stained with cynomolgus monkey cross-reactive mouse anti-human CD80 (clone L307.4) and mouse anti-human CD86 (clone 2331/FUN-1) antibodies (BD Biosciences). Molecules of equivalent soluble fluorochrome (MESF) units on CD20-gated B cells (anti-CD20 clone L27, BD Biosciences) were obtained using standard beads from Bangs Laboratories. Receptor occupancy was expressed as percent of free CD80 or CD86, with a value of 100% assigned to Day 0 (pre-dose). To assess immune responses in monkeys, the amount of anti-KLH IgG and IgM in serum was determined using monkey anti-KLH IgG1 and IgM ELISA kits from Life Diagnostics. To quantify drug levels, a DELFIA based immunoassay was developed using a goat anti-CTLA4 antibody (R&D Systems) as capture reagent and a rabbit anti-CTLA4 antibody (Novus Biologicals) for detection. Pharmacokinetic parameters for individual monkeys were determined with a non-compartmental model using Phoenix WinNonlin 6.1 (Pharsight).
Comparison of XPro9523 and belatacept in cynomolgus monkeys immunized with KLH
Female cynomolgus monkeys weighing 2.3–4.5 kg were randomized by weight, divided into 7 groups (n = 4), and given a single 1 h i.v. infusion of PBS or 0.1 mg/kg, 0.5 mg/kg, or 2.0 mg/kg of XPro9523 or belatacept in a dose volume of 0.2 or 1.0 ml/kg. KLH (3 mg in 1 ml) was administered intramuscularly 24 h after the end of infusion and a boost was given after 16 d. Pharmacokinetic and pharmacodynamic analyses, CD80 and CD86 receptor occupancy, and anti-KLH responses were determined as described above.
Pharmacokinetic simulation of human dosing of XPro9523, abatacept, and belatacept
Two-compartment models were developed for human dosing of XPro9523, abatacept, and belatacept. Half-lives and volumes of distribution for abatacept (13.1 d, 70 ml/kg) and belatacept (9.8 d, 110 ml/kg) were drawn from the full prescribing information for each drug.Citation24,Citation25 Volumes of the serum compartment and the α-phase half-lives were estimated at 40 ml/kg and 12 h, respectively. Simulations were performed in Phoenix WinNonlin 6.1 (Pharsight) using PK Model 7.
| Abbreviations: | ||
| APC | = | antigen-presenting cells |
| CFA | = | Complete Freund's Adjuvant |
| CIA | = | collagen-induced arthritis |
| CTLA-4 | = | Cytotoxic T-Lymphocyte Antigen 4 |
| ECD | = | extracellular domain |
| FcγR | = | Fcγ receptor |
| MEC | = | minimum effective concentration |
| MLR | = | mixed lymphocyte reaction |
| PBMC | = | peripheral blood mononuclear cell |
| RA | = | rheumatoid arthritis |
| SPR | = | surface plasmon resonance |
Additional material
Download Zip (146.3 KB)Acknowledgments
We thank Duc-Hanh Nguyen, Jonathan Jacinto, Araz Eivazi, Rumana Rashid, Sher Karki, and Cristina Bautista for technical contributions.
Disclosure of Potential Conflicts of Interest
All authors are employees of Xencor, Inc., and also hold stock options in Xencor.
References
- Chambers CA, Kuhns MS, Egen JG, Allison JP. CTLA-4-mediated inhibition in regulation of T cell responses: mechanisms and manipulation in tumor immunotherapy. Annu Rev Immunol 2001; 19:565 - 94; http://dx.doi.org/10.1146/annurev.immunol.19.1.565; PMID: 11244047
- Lafferty KJ, Prowse SJ, Simeonovic CJ, Warren HS. Immunobiology of tissue transplantation: a return to the passenger leukocyte concept. Annu Rev Immunol 1983; 1:143 - 73; http://dx.doi.org/10.1146/annurev.iy.01.040183.001043; PMID: 6443557
- Lafferty KJ, Woolnough J. The origin and mechanism of the allograft reaction. Immunol Rev 1977; 35:231 - 62; http://dx.doi.org/10.1111/j.1600-065X.1977.tb00241.x; PMID: 330389
- Walker LS, Sansom DM. The emerging role of CTLA4 as a cell-extrinsic regulator of T cell responses. Nat Rev Immunol 2011; 11:852 - 63; http://dx.doi.org/10.1038/nri3108; PMID: 22116087
- Wing K, Yamaguchi T, Sakaguchi S. Cell-autonomous and -non-autonomous roles of CTLA-4 in immune regulation. Trends Immunol 2011; 32:428 - 33; http://dx.doi.org/10.1016/j.it.2011.06.002; PMID: 21723783
- Linsley PS, Brady W, Urnes M, Grosmaire LS, Damle NK, Ledbetter JA. CTLA-4 is a second receptor for the B cell activation antigen B7. J Exp Med 1991; 174:561 - 9; http://dx.doi.org/10.1084/jem.174.3.561; PMID: 1714933
- Vincenti F. Costimulation blockade in autoimmunity and transplantation. J Allergy Clin Immunol 2008; 121:299 - 306, quiz 307-8; http://dx.doi.org/10.1016/j.jaci.2008.01.002; PMID: 18269922
- Larsen CP, Pearson TC, Adams AB, Tso P, Shirasugi N, Strobert E, et al. Rational development of LEA29Y (belatacept), a high-affinity variant of CTLA4-Ig with potent immunosuppressive properties. Am J Transplant 2005; 5:443 - 53; http://dx.doi.org/10.1111/j.1600-6143.2005.00749.x; PMID: 15707398
- Davis PM, Abraham R, Xu L, Nadler SG, Suchard SJ. Abatacept binds to the Fc receptor CD64 but does not mediate complement-dependent cytotoxicity or antibody-dependent cellular cytotoxicity. J Rheumatol 2007; 34:2204 - 10; PMID: 17787038
- World Health Organization. Recommended International Nonproprietary Names: List 59. WHO Drug Information 2008; 22:41 - 76
- Leister KJ, Schaefer EJ, Bates RC, Bramhall EA, Didio DM, Donaldson R, et al. (Bristol-Myers Squibb Company). Compositions and methods for producing a composition. United States Patent and Trademark Office 2009; US 20090252749A1.
- Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol 2007; 7:715 - 25; http://dx.doi.org/10.1038/nri2155; PMID: 17703228
- Zalevsky J, Chamberlain AK, Horton HM, Karki S, Leung IW, Sproule TJ, et al. Enhanced antibody half-life improves in vivo activity. Nat Biotechnol 2010; 28:157 - 9; http://dx.doi.org/10.1038/nbt.1601; PMID: 20081867
- Kuo TT, Aveson VG. Neonatal Fc receptor and IgG-based therapeutics. MAbs 2011; 3:422 - 30; http://dx.doi.org/10.4161/mabs.3.5.16983; PMID: 22048693
- Linsley PS, Greene JL, Brady W, Bajorath J, Ledbetter JA, Peach R. Human B7-1 (CD80) and B7-2 (CD86) bind with similar avidities but distinct kinetics to CD28 and CTLA-4 receptors. Immunity 1994; 1:793 - 801; http://dx.doi.org/10.1016/S1074-7613(94)80021-9; PMID: 7534620
- Xu Z, Juan V, Ivanov A, Ma Z, Polakoff D, Powers DB, et al. Affinity and cross-reactivity engineering of CTLA4-Ig to modulate T cell costimulation. J Immunol 2012; 189:4470 - 7; http://dx.doi.org/10.4049/jimmunol.1201813; PMID: 23018459
- Schwartz JC, Zhang X, Fedorov AA, Nathenson SG, Almo SC. Structural basis for co-stimulation by the human CTLA-4/B7-2 complex. Nature 2001; 410:604 - 8; http://dx.doi.org/10.1038/35069112; PMID: 11279501
- Stamper CC, Zhang Y, Tobin JF, Erbe DV, Ikemizu S, Davis SJ, et al. Crystal structure of the B7-1/CTLA-4 complex that inhibits human immune responses. Nature 2001; 410:608 - 11; http://dx.doi.org/10.1038/35069118; PMID: 11279502
- Kliwinski C, Kukral D, Postelnek J, Krishnan B, Killar L, Lewin A, et al. Prophylactic administration of abatacept prevents disease and bone destruction in a rat model of collagen-induced arthritis. J Autoimmun 2005; 25:165 - 71; http://dx.doi.org/10.1016/j.jaut.2005.09.020; PMID: 16256307
- Knoerzer DB, Karr RW, Schwartz BD, Mengle-Gaw LJ. Collagen-induced arthritis in the BB rat. Prevention of disease by treatment with CTLA-4-Ig. J Clin Invest 1995; 96:987 - 93; http://dx.doi.org/10.1172/JCI118146; PMID: 7543497
- Finck BK, Linsley PS, Wofsy D. Treatment of murine lupus with CTLA4Ig. Science 1994; 265:1225 - 7; http://dx.doi.org/10.1126/science.7520604; PMID: 7520604
- Dall’Acqua WF, Woods RM, Ward ES, Palaszynski SR, Patel NK, Brewah YA, et al. Increasing the affinity of a human IgG1 for the neonatal Fc receptor: biological consequences. J Immunol 2002; 169:5171 - 80; PMID: 12391234
- Levisetti MG, Padrid PA, Szot GL, Mittal N, Meehan SM, Wardrip CL, et al. Immunosuppressive effects of human CTLA4Ig in a non-human primate model of allogeneic pancreatic islet transplantation. J Immunol 1997; 159:5187 - 91; PMID: 9548454
- Bristol-Myers Squibb. ORENCIA (package insert). Princeton, New Jersey, USA, 2011.
- Bristol-Myers Squibb. NULOJIX (package insert). Princeton, New Jersey, USA, 2011.
- Genovese MC, Covarrubias A, Leon G, Mysler E, Keiserman M, Valente R, et al. Subcutaneous abatacept versus intravenous abatacept: a phase IIIb noninferiority study in patients with an inadequate response to methotrexate. Arthritis Rheum 2011; 63:2854 - 64; http://dx.doi.org/10.1002/art.30463; PMID: 21618201
- Bristol-Myers Squibb. Belatacept (BMS-224818): briefing document for March 2010 meeting (BLA 125288). FDA Cardiovascular and Renal Drugs Advisory Committee, 2010.
- Kirk AD, Harlan DM, Armstrong NN, Davis TA, Dong Y, Gray GS, et al. CTLA4-Ig and anti-CD40 ligand prevent renal allograft rejection in primates. Proc Natl Acad Sci U S A 1997; 94:8789 - 94; http://dx.doi.org/10.1073/pnas.94.16.8789; PMID: 9238056
- Moreland LW, Alten R, Van den Bosch F, Appelboom T, Leon M, Emery P, et al. Costimulatory blockade in patients with rheumatoid arthritis: a pilot, dose-finding, double-blind, placebo-controlled clinical trial evaluating CTLA-4Ig and LEA29Y eighty-five days after the first infusion. Arthritis Rheum 2002; 46:1470 - 9; http://dx.doi.org/10.1002/art.10294; PMID: 12115176
- Brand DD, Latham KA, Rosloniec EF. Collagen-induced arthritis. Nat Protoc 2007; 2:1269 - 75; http://dx.doi.org/10.1038/nprot.2007.173; PMID: 17546023