
Abstract
Limited access for high-quality biologics due to cost of treatment constitutes an unmet medical need in the United States (US) and other regions of the world. The term “biosimilar” is used to designate a follow-on biologic that meets extremely high standards for comparability or similarity to the originator biologic drug that is approved for use in the same indications. Use of biosimilar products has already decreased the cost of treatment in many regions of the world, and now a regulatory pathway for approval of these products has been established in the US. The Food and Drug Administration (FDA) led the world with the regulatory concept of comparability, and the European Medicines Agency (EMA) was the first to apply this to biosimilars. Patents on the more complex biologics, especially monoclonal antibodies, are now beginning to expire and biosimilar versions of these important medicines are in development. The new Biologics Price Competition and Innovation Act allows the FDA to approve biosimilars, but it also allows the FDA to lead on the formal designation of interchangeability of biosimilars with their reference products. The FDA’s approval of biosimilars is critical to facilitating patient access to high-quality biologic medicines, and will allow society to afford the truly innovative molecules currently in the global biopharmaceutical industry’s pipeline.
Note
This paper is based on presentations given by Mark McCamish on November 30, 2010 at the 6th European Antibody Congress in Geneva, Switzerland;Citation24 on December 8, 2010 at IBC's 8th Annual International Conference on Antibody Therapeutics held in San Diego, CA and also on the presentation and submission made by the Novartis Group of companies to the FDA Docket on Approval Pathway for Biosimilar and Interchangeable Biological Products.Citation18
Appendix
Definitions.
Biosimilar, PHSA 351(k) application (US) or “similar biological medicinal products” (EU); BPCIA, Biologics Price Competition and Innovation Act, the biosimilars provisions of the US health care reform legislation enacted on March 23, 2010); EMA, European Medicines Agency, formerly known as the European Medicines Evaluation agency (EMEA); Hatch Waxman act, Drug Price Competition and Patent Term Restoration act of 1984, also known as Hatch Waxman; FFDCA, Federal Food, Drug and Cosmetic act, the statute under which most drugs have been approved in the US; PHSA, Public Health Service Act, the statute under which most biologics have been licensed (approved) in the US.
Figures and Tables
Figure 1 European biosimilar guidelines. The EMA began with an overarching guideline on biosimilars and then general guidelines, before issuing product class specific data requirements. The EU Guidelines that have been finalized are indicated in blue. A draft guideline for mAbs is currently available for public comment.Citation25
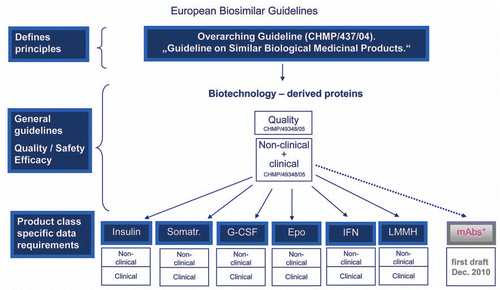
Figure 2 Analyzing complex product attributes over time. The bG2 glycan structure was quantified by Sandoz in many batches of commercial product distributed by the originator in the EU (light blue) and the US (dark blue). Expiry date of the product batches is listed on the x-axis and relative amount of product attribute enrichment is listed on the y-axis. Pre-shift quality refers to the content of the attribute prior to a manufacturing change and post-shift quality after the manufacturing change.
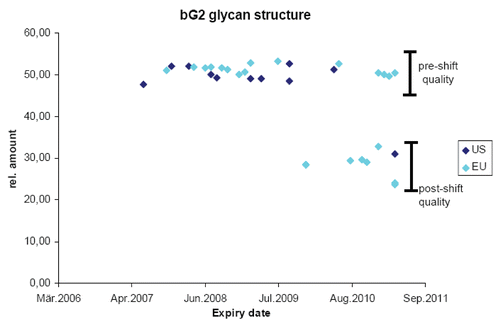
Figure 3 Biosimilar development process. The development of a biosimilar relies on creation of a design space based on analysis of the reference product and then iterative development of a biosimilar to fit the chosen specifications. There is no access to, nor need for, originator data at any point in this process. The early process development is essential, and later development cannot compensate for this initial generation of a “highly similar” candidate product. As the complexity of the reference product increases, the initial development becomes more challenging, and the likelihood that multiple iterations will be needed increases.
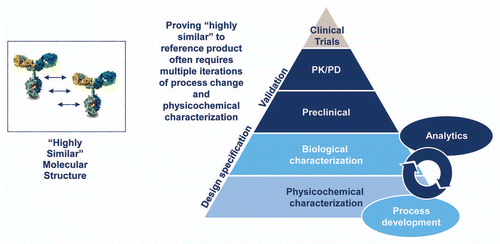
Figure 4 Biosimilarity goal posts. The “goal posts” of biosimilarity are established by the biosimilar sponsor by their analysis of the distribution of product attributes present in the reference product pre- and post-manufacturing change. They then use these to select the design space for their biosimilar candidate. While the complete quality range may be quite broad for the life time of the reference product, the biosimilar sponsor will select a tighter range of control for their biosimilar product.
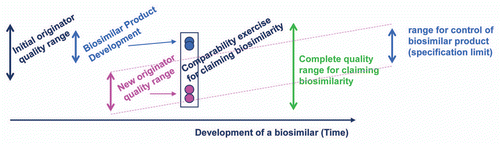
Figure 5 The one US marketed generic biologic: enoxaparin. Generic enoxaparin was approved by FDA on July 23, 2010. Although not a “biosimilar” insofar as it was not approved through the new biosimilar 351(k) regulatory pathway, scientifically it is a fully interchangeable generic biologic and many of the FDA considerations anticipated for biosimilars s apply.

Notes
For explanation of abbreviations, see Appendix.