Abstract
Off-target binding can significantly affect the pharmacokinetics (PK), tissue distribution, efficacy and toxicity of a therapeutic antibody. Herein we describe the development of a humanized anti- fibroblast growth factor receptor 4 (FGFR4) antibody as a potential therapeutic for hepatocellular carcinoma (HCC). A chimeric anti FGFR4 monoclonal antibody (chLD1) was previously shown to block ligand binding and to inhibit FGFR4 mediated signaling as well as tumor growth in vivo. A humanized version of chLD1, hLD1.vB, had similar binding affinity and in vitro blocking activity, but it exhibited rapid clearance, poor target tissue biodistribution and limited efficacy when compared to chLD1 in a HUH7 human HCC xenograft mouse model. These problems were traced to instability of the molecule in rodent serum. Size exclusion high performance liquid chromatography, immunoprecipitation and mass spectral sequencing identified a specific interaction between hLD1.vB and mouse complement component 3 (C3). A PK study in C3 knock-out mice further confirmed this specific interaction. Subsequently, an affinity-matured variant derived from hLD1.vB (hLD1.v22), specifically selected for its lack of binding to mouse C3 was demonstrated to have a PK profile and in vivo efficacy similar to that of chLD1 in mice. Although reports of non-specific off-target binding have been observed for other antibodies, this represents the first report identifying a specific off-target interaction that affected disposition and biological activity. Screens developed to identify general non-specific interactions are likely to miss the rare and highly specific cross-reactivity identified in this study, thus highlighting the importance of animal models as a proxy for avoiding unexpected clinical outcomes.
Antibodies are an attractive source of biotherapeutic agents due to their high affinity, exquisite target selectivity and extended half-life in vivo. Their development for therapeutic applications has been facilitated by hybridoma technology, antibody humanization and numerous in vitro antibody selection technologies that enable antibodies with desired biological properties to be engineered at will.
Co-incident with this increase in the in vitro development of antibodies for therapeutic applications has been the recognition of how antibodies evolve in vivo. Several studies have pointed to the relationship between antibody affinity and antigen specificity.Citation1–Citation6 The conformational flexibility of initial recombined antibodies is considered to be an important feature of the immune system's ability to generate antibodies against a broad spectrum of antigens. During antibody maturation, this structural plasticity is thought to be restricted through somatic hypermutation in vivo (and perhaps affinity maturation in vitro) leading to a reduced entropy cost for specific antigen binding and a corresponding increase in antigen specificity. The increase in antigen specificity helps to eliminate undesired off-target antibody interactions, and serves as part of immune system checkpoints designed to prevent autoimmune disease.
In contrast, antibodies that are generated in vitro lack any regulatory immune surveillance. For these, various screens utilizing protein chips and microarrays have been developed in order to evaluate or anticipate off-target interactions.Citation7–Citation9 In one study comparing antibodies against TNFα, for example, multiple off-target interactions were found for adalimumab, an antibody derived in vitro from a cloned human antibody phage library.Citation7 However, such screens are artificial and whether any observed non-specific off-target binding events result in adverse side effects in vivo or actually take place in vivo has yet to be demonstrated.
Recently, affinity matured variants of palivizumab were found to have a less than anticipated increase in potency as a prophylactic treatment in a rat model of respiratory syncytial virus (RSV). The variants unexpectedly exhibited broad tissue binding that led to their rapid clearance and low target tissue bioavailability.Citation10 Reversion of some of the amino acid changes incorporated during affinity maturation diminished much of the non-specific tissue binding and improved efficacy and pharmacokinetics. Direct interactions leading to the broad non-specific tissue binding were thus identified and removed, enabling the development of a more effective variant of palivizumab. This is the first report correlating broad tissue cross-reactivity in vitro with rapid clearance and poor tissue bioavailability.
The likelihood of identifying an off-target binding event is a function of the size of the protein repertoire and the affinity that is considered relevant.Citation11 Given the huge complexity of an in vivo system and the typically high therapeutic dosing in a clinical setting, the odds of an off-target event that affects efficacy, clearance, tissue bioavailability or toxicity are greatly increased. Such antigen promiscuity in an antibody may arise from the recognition of structurally related epitopes (molecular mimicry), the utilization of overlapping or independent antibody paratopes or through conformational flexibility that enables the complementary-determining regions (CDRs) or side chains of an antibody to adapt to more than one antigen.Citation12,Citation13
In contrast to the broad non-specific off-target recognition and antibody polyspecificity described above, here we report an unexpected specific off-target binding event we identified for a humanized antibody directed toward fibroblast growth factor receptor (FGFR) 4. The FGFR signaling system plays critical roles in a variety of normal developmental and physiological processes, and aberrant signaling may lead to tumor development and progression.Citation14 FGFR4 has been shown to play a modulatory role in the development and progression of hepatocellular carcinoma (HCC) in mice, and potentially in humans. A chimeric anti-FGFR4 monoclonal antibody (chLD1) was previously shown to block ligand binding, inhibit FGFR4-mediated signaling and inhibit HCC tumor growth in vivo.Citation14 Following the humanization of chLD1, we observed an unexpected loss of efficacy in a mouse tumor model, although the humanized variant had identical affinity for FGFR4. This variant was found to cross-react with an abundant mouse serum protein. This specific off-target interaction interfered with FGFR4 binding, altered antibody clearance, impacted target tissue distribution, resulting in reduced therapeutic activity. Subsequently we eliminated this off-target binding through affinity maturation of the humanized antibody leading to the full restoration of the in vivo properties inherent in chLD1.
This work illustrates some of the challenges that extend well beyond simple antibody-antigen binding and serves as a cautionary tale to therapeutic antibody development.
Results
Humanization and characterization of LD1.
The human chimeric antibody LD1 (chLD1) has been shown to bind human FGFR4, block signaling by FGF19 and other FGF ligands and suppress tumor growth in a HUH7 human hepatocelluar carcinoma (HCC) xenograft model.Citation14 As a first step in the humanization of LD1, the variable light and heavy domains of chLD1 were aligned with the human kappa I (huKI) and human VH subgroup III (huIII) variable domain frameworks used in trastuzumab (). Hypervariable regions from chLD1 were grafted into these human variable frameworks to generate a direct CDR-graft (hLD1.vA).Citation15,Citation16 When compared to chLD1 by surface plasmon resonance for binding FGFR4, the affinity of hLD1.vA was decreased by about 5-fold (not shown). Substitution of mouse sequence at various vernier positions in both the variable light and heavy domains was explored as a means to improve binding and led to the identification of three important mouse vernier positions in the LC: P44F, L46I and Y49S. These changes were incorporated in hLD1.vB that had an affinity for FGFR4 comparable to chLD1 ().
Surprisingly, despite similar FGFR4 binding affinity (), hLD1.vB had diminished anti-tumor efficacy compared to chLD1 in the HUH7 human HCC xenograft model in nu/nu mouse (). After 9 days, the HUH7 tumors of mice treated with PBS grew to an average volume of approximately 700 mm3. In the chLD1 treated group, average HUH7 tumor volume was approximately 400 mm3, representing a 43% inhibition of tumor growth compared to the tumor in the PBS-treated animals. However, the average tumor volume in mice treated with hLD1.vB was approximately 600 mm3, representing a 14% inhibition of tumor growth compared to the tumor in the PBS-treated animals.
A pharmacokinetic evaluation of chLD1 and hLD1.vB conducted in athymic NCR nude mice revealed rapid clearance for both chLD1 and hLD1.vB at 1 mg/kg IV (140 and 132 mL/day/kg, respectively), suggesting a target mediated clearance mechanism. This clearance mechanism appeared to be saturated for chLD1 at a higher dose of 20 mg/kg. At this dose the observed clearance (11.7 mL/day/kg; ) was within the range (6–12 mL/day/kg) of target-independent clearance observed for a typical humanized antibody in mouse (ref. Citation17; P. Theil, personal communication). However, hLD1.vB continued to be rapidly cleared (34.2 mL/day/kg; ). This suggested an additional clearance mechanism for hLD1.vB could be responsible for the apparent lack of efficacy in the mouse xenograft model.
Consistent with the pharmacokinetics (PK) finding, a biodistribution study using 125I-chLD1 and 125I-hLD1.vB revealed significantly different distribution profiles (). 125I-chLD1 distributed rapidly and specifically to the liver due to the high expression of FGFR4 on hepatocytes while only a limited amount of 125I-hLD1.vB was found in liver at an equivalent dose by 2 h (∼80 vs. 35% ID/g). In contrast, the observed distribution of these antibodies was reversed in blood suggesting that a competing interaction prevented distribution of hLD1.vB to the liver as opposed to a loss in antibody stability in vivo that would have led to a loss in overall radioactivity.
Identification of C3 interference.
In an effort to reconcile the in vivo differences observed between chLD1 and hLD1.vB, we evaluated antibody stability in plasma as well as potential off-target plasma or tissue interactions that might affect their function. Plasma stability was evaluated by incubating chLD1 or hLD1.vB in mouse, rat, monkey or human plasma for 48 hours at 37°C followed by an assessment of both the FGFR4 binding activity and the total human IgG concentration. While the total chLD1 or hLD1.vB concentration as measured by the IgG ELISA did not change (not shown), the recovery of hLD1.vB detected by the FGFR4 ELISA was significantly reduced (by ∼30%) in mouse and rat plasma compared to a control incubation in PBS/BSA (). In contrast, there was no loss of chLD1 FGFR4 binding activity in any condition tested. The significant reduction in hLD1.vB recovery, specifically from rodent plasma, suggested that the loss was not due to degradation, but more likely the formation of an interfering complex in rodent plasma. Since the interaction of hLD1.vB with mouse plasma might result in the generation of a higher molecular weight complex, iodinated chLD1 and hLD1.vB were also incubated in plasma and analyzed using size-exclusion HPLC. High molecular weight peaks were detected only in the mouse plasma samples containing 125I-hLD1.vB but not 125I-chLD1. In addition to the expected antibody peak at 150 kDa, peaks corresponding to ca. 270 and ca. 550 kDa were also detected initially (), however by 48 h, only the 150 and 270 kDa peaks remained; the 550 kDa peak was no longer observed. These higher molecular weight peaks were not detected in cynomolgus monkey and human plasma or PBS/BSA containing hLD1.vB, or in any sample containing chLD1 (Sup. Fig. 1). Interestingly, the presence of these high molecular weight peaks correlated directly with the antibody recovery data obtained in the FGFR4 ELISA. Further, the presence of these peaks was diminished when the analysis was performed at pH 4.0 (Sup. Fig. 1), further supporting the hLD1.vB-dependent interaction with mouse serum.
Immunoprecipitation of mouse plasma revealed a ca. 37 kDa protein that was selectively precipitated using hLD1.vB, but not chLD1 (). Consistent with the findings from size-exclusion HPLC, this 37 kDa protein band was observed in rat but not cynomolgus monkey and human plasma samples (Sup. Fig. 2A–C). Furthermore, the protein was detected in the plasma from mice administered hLD1.vB (Sup. Fig. 2D). MS/MS analysis of tryptic peptides derived from the 37 kDa mouse plasma protein identified this band as being derived from mouse complement C3 (). A direct involvement of C3 was supported by the full recovery of hLD1.vB incubated in plasma from C3 knock out (ko) mice (Sup. Fig. 3).
Affinity maturation and re-evaluation of C3 binding.
Both chLD1 and hLD1.vB share the same human constant regions and CDRs and thus differ only by their variable domain frameworks. Further, the light and heavy chain variable domain frameworks used to humanize hLD1.vB share a very high degree of homology with several humanized antibodies, including trastuzumab that have not been reported to exhibit interactions with mouse serum proteins. The off-target interaction of hLD1.vB with mouse C3 thus most likely resulted from the particular combination of the mouse LD1 CDRs with the human variable domain frameworks.
We reasoned that some of the changes in the CDR sequences of hLD1.vB, which resulted from affinity maturation of the Fab fragment displayed on phage, could lead to improved affinity for FGFR4 with a concomitant loss in mouse C3 binding. Phage-selected variants were expressed as IgG and screened for FGFR4 binding affinity, as well as potential interaction with mouse C3, using an immunoprecipitation assay coupled with SDS-PAGE analysis. One variant, hLD1.v22, with 3 amino acid changes in CDR-H2 compared to hLD1.vB (H52L, S53V and D60E, ), showed both improved binding affinity for FGFR4 () and a loss of binding to complement C3 ().
The degree to which mouse complement C3 altered the clearance of hLD1.vB and hLD1.v22 in vivo was assessed in a PK study comparing C3 ko mice to C3 wild type (wt) mice at 20 mg/kg. As observed previously in NCR mice, hLD1.vB was rapidly cleared from circulation in C3 wt mice (29 mL/day/kg); however, both chLD1 and hLD1.vB had similar PK profiles (clearance of 8.7 and 9.3 mL/day/kg, respectively) in the C3 ko mice ( and Sup. Table 1). These data are consistent with the tissue distribution data and confirm that specific interaction with mouse complement C3 in vivo leads to rapid clearance of hLD1.vB. Since the clearance of hLD1.vB is significantly improved in C3 ko mice and hLD1.v22 does not bind to C3, we anticipated that hLD1.v22 would have a PK profile similar to chLD1 in NCR nude mice. As shown in , the clearance of chLD1 and hLD1.v22 are similar in NCR nude mice (11.8 and 11.3 mL/day/kg respectively) while consistent with our previous findings, hLD1.vB is rapidly cleared (46.7 mL/day/kg).
The ability of hLD1.v22 to inhibit tumor growth in the HUH7 HCC xenograft model was evaluated in comparison to chLD1 and hLD1.vB. After 21 days, the HUH7 tumors of mice treated with PBS grew to an average volume of approximately 2,100 mm3 (). Of the 15 animals in the PBS-treated group, three were euthanized before the end of study due to tumor volume limits (approximately 2,500 mm3). In the hLD1.vB treated group, average HUH7 tumor volume was approximately 1,200 mm3, representing a 43% inhibition of tumor growth compared to the tumors in PBS-treated animals. However, the average tumor volume in mice treated with hLD1.v22 was approximately 530 mm3. This result was comparable to the chLD1 treated mice where the average HUH7 tumor volume was approximately 350 mm3. For both hLD1.v22 and chLD1 treated groups, this represents a 75% and 83% reduction in tumor size, respectively, compared to the PBS vehicle treatment group. The tumor doubling times of the groups treated with hLD1.vB (12.2 days), hLD1.v22 (15.8 days) or chLD1 (17.1 days) is significantly greater than that of the PBS-treated group (8.2 days). Additionally, the tumor doubling times of the hLD1.v22 or chLD1-treated groups were significantly longer than that for the hLD1.vB treated group. The similar in vivo performance of both hLD1.v22 and chLD1 compared to hLD1.vB, strongly implies that the specific off-target interaction with mouse complement C3 was causing the increased clearance resulting in lower exposure and reduced efficacy of hLD1.vB.
Discussion
As biotherapeutics, antibodies represent a promising class of molecules that offer high affinity, slow clearance and a high degree of specificity. With the advent of antibody engineering, the immune system can be bypassed thus enabling generation of a vast diversity of antibodies with novel properties. As a result, many therapeutically useful antibodies against human antigens have been generated as humanized antibodies derived from mouse hybridoma technology or selected directly using cloned or synthetic antibody repertories expressed on phage and other display formats.Citation18
Despite the power of the in vitro approach, engineered antibodies have an Achilles heel. Antibodies made in vitro suffer from the lack of in vivo selection and regulatory mechanisms. Host-generated antibodies that cross-react with self-antigens or interact broadly with normal tissues are normally eliminated to prevent autoimmune disease. In contrast, in vitro engineered molecules are not surveyed by this immune regulatory selection process.
In this work, we encountered an unexpected rapid clearance of a humanized antibody that was absent in the originating chimera. Although chLD1 and hLD1.vB had similar affinity and equally inhibited ligand binding to FGFR4 in vitro ( and ), hLD1.vB had diminished anti-tumor efficacy in vivo compared to chLD1 in the HUH7 HCC xenograft mouse model (). A tissue distribution study comparing chLD1 and hLD1.vB revealed rapid and specific distribution of chLD1 to liver, a site with high FGFR4 expression, while hLD1.vB remained primarily in circulation (). In addition, the recovery of hLD1.vB added to mouse or rat plasma in vitro was significantly reduced compared to the recovery of chLD1 or hLD1.vB added to plasma from cynomolgus monkey or human (). Analysis of mouse and rat serum samples by size exclusion HPLC revealed the presence of high molecular weight complexes specific to rodent serum samples containing hLD1.vB ( and Sup. Fig. 1). While the rapid clearance of hLD1. vB might be explained by potential plasma instability, aggregation or off-target binding, the lack of any low molecular weight peaks suggested degradation was unlikely and the lack of high molecular weight peaks in cynomolgus monkey and human samples, reduced the probability of aggregation, as one would expect aggregation to be species independent. Thus the generation of an hLD1.vB-plasma protein complex specifically in rodents seemed likely.
Immunoprecipitated samples corresponding to these high molecular weight peaks revealed an unexpected 37 kDa band in addition to bands corresponding to hLD1.vB. Mass spectral sequencing of peptides generated from the 37 kDa band indicated they were derived from mouse complement C3 and localized around a specific region encoding C3d ( and Sup. Fig. 2). Complement C3 is highly abundant in serum. Synthesized by hepatocytes, monocytes, macrophages and astrocytes, it normally plays an important role in identifying and clearing immune complexes from blood.Citation19,Citation20 Activation of C3 (185 kDa) by C3 convertases leads to the formation of C3b followed by further processing to form C3d, a 35 kDa proteolytic fragment of C3b. Although C3 fragments can form spontaneously within the circulation in vivo or immediately following blood draw and during ex vivo incubations, C3 fragments are not detected in circulation until complete blood stasis is attained. This is probably due to rapid clearance mechanisms or because the normal in vivo hydrolysis of C3 is thought to occur quite slowly.Citation20–Citation22 The 550 and 270 kDa peaks observed by size exclusion HPLC are consistent with the formation over time of an initial complex composed of two C3 molecules bound to hLD1.vB (ca. 520 kDa) followed by the conversion to two C3d molecules bound to hLD1.vB (ca. 220 kDa) over time (). A direct role for mouse C3 in the clearance of hLD1.vB in NCR mice was supported by the improved serum recovery () and normal clearance of hLD1.vB relative to chLD1 in C3 ko mice ().
Conversion of C3 to C3b results in the formation of a covalent thioester bond between C3b and its target proteins such as IgG. This thioester bond originates in the C3d region and links to IgG through the CH1 constant domain.Citation23,Citation24 The interaction of hLD1.vB with C3d in this instance appears to be exclusively through a unique non-covalent interaction as the observed complex was completely dissociated at lower pH and by SDS-PAGE ( and Sup. Fig. 1). In addition, the association of hLD1.vB with C3d only occurs in rodents, not in primates, despite the human constant domain usage in hLD1.vB. Finally, the interaction appears to involve the variable domains of hLD1.vB, since in serum, the presence of C3/C3d appears to block the interaction with FGFR4 ( and ).
The decrease in the ability of hLD1.vB to bind FGFR4 observed after incubation in rodent serum () suggests that at equilibrium only about 25% of the 1.3 µM hLD1.vB added is in complex with complement C3 despite the high C3 abundance (150 µg/ml or 0.8 µM) in mouse serum.Citation25 This is indicative of a relatively weak interaction between hLD1.vB and mouse C3, although the impact of this interaction is significant in vivo. The generation of this C3/C3d-hLD1.vB complex appears to block interaction with FGFR4, cause rapid clearance and prevent distribution to the liver; all leading to a loss of efficacy in the HUH7 human HCC mouse xenograft model ().
Although the C3/C3d-hLD1.vB complex is likely a limited problem specific to rodents, preclinical evaluation in a mouse xenograft model is often a necessary step in therapeutic antibody development. To eliminate this unintended interaction, we examined the differences between chLD1 and hLD1.vB (). The interaction with C3 cannot be ascribed to the CDR sequences since they are identical to that of chLD1. Further, the human variable light and heavy domain framework sequences are essentially identical to those used in many other humanized antibodies. Surprisingly, these features, when combined in the form of hLD1.vB, led to an interaction with mouse C3 and this interaction also blocked binding to FGFR4. Thus, if the interaction with C3 was dependent upon a combination between the CDR sequences from chLD1 and the human variable light and heavy domain frameworks (), we reasoned that amino acid changes in the CDRs obtained through affinity maturation of hLD1.vB on FGFR4 might disrupt C3 binding. Using serum recovery in the FGFR4 ELISA and immunoprecipitation as in vitro screens, we selected an affinity-matured variant of hLD1.vB, hLD1.v22, which appeared to be stable in mouse serum and did not immunoprecipitate C3d (). Subsequent in vivo studies demonstrated that the clearance and efficacy of hLD1.v22 was similar to that observed for chLD1 in normal mice (). The three amino acid changes in hLD1.v22, located in CDR-H2 of the heavy chain, both improve the affinity for FGFR4 () and eliminate the interaction with mouse C3.
Recently, Wu et al. also described an affinity-matured antibody variant derived from palivizumab that exhibited rapid clearance and limited bioavailability.Citation10 Subsequent investigations revealed broad non-specific tissue binding as determined by immunohistochemistry on frozen tissue samples that resulted from specific mutations performed during affinity maturation of palivizumab. The authors report that reversion of three amino acid residues, introduced during the affinity maturation process, back to the original palivizumab sequence, led to a reduction in non-specific tissue binding and improved overall exposure and efficacy in vivo. In contrast to the observations of Wu et al. where the affinity maturation of palivizumab led to a broad non-specific tissue binding, this study reveals a specific off-target binding interaction between mouse C3 and hLD1.vB and the subsequent elimination of this interaction through affinity maturation. Affinity maturation thus resulted in an increase of non-specific off-target binding for palivizumab, but the loss of a specific off-target binding for hLD1.vB.
In contrast to polyreactive or multispecific antibodies typically associated with a much broader range of off-target antibody binding,Citation1,Citation3,Citation7,Citation10,Citation26 this is the first reported instance of an antibody acquiring an unintended but highly specific off-target interaction. Several lines of evidence suggest that in early antibody development, polyreactive B-cells play an important role for providing a diverse antibody repertoire that can be recruited for the generation of high affinity antibodies against a diverse population of antigens. This multi-specificity is thought to result from conformational flexibility of the CDRs. During antibody affinity maturation through somatic mutation in vivo, this flexibility is thought to be reduced leading to increased affinity and specificity.Citation4–Citation6
With this in mind, one possibility is that conformational flexibility may have been introduced upon grafting the LD1 CDRs into the new human framework such that new interactions with C3d were enabled. Thus, rather than altering a direct interaction with C3 through affinity maturation, the changes made to CDR-H2 may have restricted the flexibility of this loop in a way that limited its interaction to FGFR4. The question of whether or not changes in CDR-H2 alter CDR flexibility or interact directly with FGFR4 will require structural conformation; however, from a kinetic standpoint, reduced flexibility would likely be reflected in an increased association rate. Compared to hLD1.vB, the association rate of hLD1.v22 was increased by 3-fold but this was only a minor component of the overall increase in affinity of 20-fold. We suspect therefore that the particular combination of LD1 CDR and human variable domain sequences led to the weak but specific interaction with mouse C3. These types of events are likely to be difficult to predict or eliminate in antibody development screening processes.
In the context of clinical development, toxicity resulting from off-target binding is an overriding concern. A therapeutic candidate that exhibits general tissue cross-reactivity might be flagged by in vitro screens such as protein microarrays,Citation7,Citation9,Citation27,Citation28 light directed peptide synthesis arraysCitation29 and tissue cross-reactivity assays.Citation30–Citation32 These screens along with preclinical evaluation in rodents and primates are important tools in the assessment of how well a molecular will behave in the clinic. But how can the risk of rare species-dependent highly specific off-target interactions be safe-guarded? Further, the evidence leading to the identification of mouse C3 was facilitated by the presence of off-target binding in the blood compartment, whereas off-target binding may just as easily occur in a particular organ or tissue. For clinical trials that fail due to unexplained toxicity or lack of efficacy, one may be left wondering if there might be a unique off-target interaction that had previously gone unnoticed.
Materials and Methods
FGFR4 was biotinylated using Sulfo-NHS-LC-biotin (Pierce; cat. 21335).
Generation of chimeric LD1.
The extracellular domain of human FGF receptor 4 (FGFR4), expressed in CHO cells and purified as described in reference Citation14, was used to immunize balb/c mice. A hybridoma expressing LD1 was identified when clones were screened for the ability to block FGF19 binding to FGFR4 in a protein-based ELISA.
The murine LD1 variable domains were cloned from total RNA extracted from LD1 producing hybridoma cells using standard methods. The variable light (VL) and variable heavy (VH) domains were amplified using RT-PCR with degenerate reverse primers to the constant light (CL) and constant heavy domain 1 (CH1) and forward primers specific for the N-terminal amino acid sequence of the VL and VH regions. These variable domains were then cloned in-frame into vectors that contained the respective human light and heavy chain constant regions.
Humanization and affinity maturation of LD1.
Hypervariable regions of LD1 were grafted into the human kappa I (huKI) and human VH subgroup III (huIII) variable domain frameworks used in trastuzumab. Framework repair was used to optimize FGFR4 binding affinity through the addition of mouse vernier position until an minimum combination of framework changes was identified that fully restored FGFR4 binding affinity.Citation33
hLD1.vB, displayed as a monovalent Fab-P3 fusion on phage, was affinity matured using a soft randomization strategy. Sequence diversity was introduced separately into each hypervariable region such that a bias towards the murine hypervariable region sequence was maintained using a poisoned oligonucleotide synthesis strategy.Citation34 For each diversified position, the codon encoding the wild-type amino acid is poisoned with a 70-10-10-10 mixture of nucleotides resulting in an average 50% mutation rate at each position.
The hLD1.vB diversified phage libraries were panned using a soluble selection method.Citation35 This approach relied upon a short binding period with low concentrations of biotinylated FGFR4 in solution, followed by a short 5 min capture of phage-bound to FGFR4 on immobilized neutravidin. Excess unlabeled FGFR4 (greater than 100 nM) was added prior to the capture step for increased off-rate selection stringency. Bound phage were eluted by incubating the wells with 100 mM HCl for 30 min, neutralized with 1 M Tris, pH 8 and amplified using XL1-Blue cells and M13/KO7 helper phage.
Phage antibodies were reformatted into full-length IgG, transiently expressed in mammalian cells and purified by Protein A chromatography.
Affinity determinations.
Affinity determinations were performed by surface plasmon resonance using a BIAcore™-2000. Approximately 50 RU of hLD1.vB IgG was immobilized in 10 mM Sodium Acetate pH 4.8 on a CM5 sensor chip and serial 2-fold dilutions of the FGFR4 (0.48–1,000 nM) in PBST were injected at a flow rate of 30 µl/min. Each sample was analyzed with 4-minute association and 10-minute dissociation. After each injection the chip was regenerated using 10 mM glycine pH 1.7. Binding response was corrected by subtracting the RU from a flow cell with an irrelevant IgG immobilized at similar density. A 1:1 Languir model of simultaneous fitting of kon and koff was used for kinetics analysis.
Xenograft experiments.
All animal protocols were approved by Genentech's Institutional Animal Care and Use Committee. Female nu/nu (nude-CRL) mice at seven weeks of age were obtained from Charles River Laboratories International (strain code 088). Mice were maintained under specific pathogen-free conditions. HUH7 cells (5 × 106; Japan Health Science Research Resources Bank, cat. JCRB0403) were implanted subcutaneously into the flank of mice in a volume of 0.2 mL in HBSS/Matrigel (1:1 v/v; BD Biosciences, cat. 354234). Tumors were measured twice weekly with a caliper and tumor volume was calculated using the formula: V = 0.5 × L × W2, where L and W are the length and width of the tumor, respectively. When the mean tumor volume reached 145 mm3, mice were randomized into groups (n = 15) and treated once weekly with 0.2 mL intraperitoneal injections of vehicle (PBS), 30 mg/kg chLD1, 30 mg/kg hLD1.vB or 30 mg/kg hLD1.v22. Following treatment, tumor volumes were measured as described above. Percent tumor growth inhibition (% TGI) was calculated using the following formula, in which C = the mean tumor volume on day 21 of the control vehicle group and T = the mean volume on day 21 from each group of mice given the test treatment: %TGI = 100 × [(C − T)/C]. Data were analyzed and log-rank tests were used to evaluate tumor doubling differences between groups with JMP software, Version 6.0 (SAS Institute). Data are presented as the mean tumor volume ± SEM.
Pharmacokinetic studies in mice.
NCR nude mice were supplied by Taconic (cat. NCRNU). C3 knock-out miceCitation36 were back-crossed to C57BL/6 mice for at least ten generations. Offspring were intercrossed to produce C3 knock-out mice and wild-type controls. In this study, they are referred to as C3 ko and C3 wt mice, respectively.
Mice weighing between 15.5 and 38.3 g were administered a 1, 5 or 20 mg/kg body weight IV bolus dose of anti-FGFR4 via the tail vein. Blood samples were collected via retro orbital bleed or cardiac stick (n = 3 mice per time point) and serum isolated at selected time points up to 28 day post dose. Serum samples were stored at −80°C until assayed for anti-FGFR4 serum concentrations using an ELISA.
Anti-FGFR4 serum concentration-time profiles were used to estimate PK parameters using WinNonlin Enterprise Version 5.2.1 (Pharsight Corp.). Since a single concentration-time profile was determined for each group, one estimate of each PK parameter was obtained and is reported, along with the standard error (SE) of the fit of each PK parameter. The nominal dose administered for each group was used for modeling.
Radioiodinations.
Antibodies were radioiodinated using an indirect iodogen addition method.Citation37 The radiolabeled proteins were purified using NAP5™ columns (GE Healthcare Life Sciences, cat. 17-0853-01) pre-equilibrated in PBS. The specific activities of molecules used in the in vitro studies were 14.38 µCi/µg for chLD1 and 15.05 µCi/µg for hLD1.vB. The specific activities of molecules used in the in vivo study were 12.52 µCi/µg for chLD1 and 9.99 µCi/µg for hLD1. vB. Following radioiodination, the radioiodinated antibodies were characterized by size-exclusion high-performance liquid chromatography (HPLC), sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and ELISA as intact and retaining comparable antigen binding to the unlabeled antibodies.
In vitro incubations.
For incubations in serum, antibodies were added to NCR nude, C3 ko or C3 wt mouse serumCitation36 and PBS + 0.5% BSA at a final concentration of 200 µg/ml. Aliquots (100 µl) were made and incubated at 37°C with gentle rotation. The samples were transferred to dry ice at 0, 4, 8, 24, 48 and 96 h and stored at −70°C until ELISA analysis.
For incubations in plasma, antibodies were added to cynomolgus monkey, human, rat (Bioreclamation LLC, cat. CYNPLLIHP, HMPLLIHP and RATPLLIHP, respectively) and NCR nude mouse plasma (Taconic, cat. NCRNU-F) and PBS + 0.5% BSA at a final concentration of 200 µg/ml ± 125I-antibodies at a final concentration of 5 × 106 CPM/ml. Aliquots (100 µl) were made and incubated at 37°C with gentle rotation. At 0, 24 and 48 h the samples were transferred to dry ice and stored at −70°C until analysis by size-exclusion HPLC (125I-antibody + unlabeled antibody samples) or subjected to protein-G extraction followed by SDS-PAGE (unlabeled antibody samples).
Tissue distribution study.
Female NCR nude mice received an IV bolus dose of 125I-chLD1 at 300 µCi/kg ± unlabeled chLD1 at 20 mg/kg or 125I-hLD1.vB at 300 µCi/kg ± unlabeled hLD1.vB at 20 mg/kg. Blood was collected at 15 minutes and 2, 5, 24, 72 and 120 h post-dose and processed for serum. The serum was frozen at −70°C until analyzed by size-exclusion HPLC and subjected to protein-G extraction followed by SDS-PAGE separation. Total radioactive counts were also obtained using a “Wallac 1480 Wizard 3” (EC&G Wallac, cat. 1480-011). Liver, lungs, kidneys and heart were collected at 2, 72 and 120 h post-dose and frozen at −70°C until analysis for total radioactivity. All animals were handled in accordance with IACUC guidelines.
IgG and FGFR4 ELISA.
FGFR4 specific IgG was measured using the FGFR4 ELISA. FGFR4 was immobilized on microtiter plates, LD1 standard (chLD1, hLD1.vB or hLD1.v22) and samples diluted in Magic Buffer + 0.35 M NaCl (1x PBS pH 7.4, 0.5% BSA, 0.05% Tween-20, 0.25% Chaps, 5 mM EDTA, 0.2% BgG, 0.35 M NaCl, 15 ppm Proclin) were added and capture IgG was detected with a F(ab')2 goat anti-huIgG Fc conjugated to horseradish peroxidase (HRP, Jackson, cat. 109-036-098).
Total antibody was determined using the Human Fc ELISA by incubating diluted samples on microtiter plates coated with F(ab')2 rabbit anti-huIgG Fc (Jackson, cat. 309-006-008) and detected with F(ab')2 goat anti-huIgG Fc conjugated to HRP (Jackson, cat. 109-036-098). TMB peroxidase substrate solution was used for color development (Moss, cat# TMBE-1000) and 1 M H3PO4 was added to stop the reaction. Plates were read on a microplate reader (Biotek EL311 or equivalent) at 450/620 nm.
Size-exclusion HPLC.
Size-exclusion HPLC was performed in PBS using a Phenomenex™ BioSep-SEC-S 3000 column 300 × 7.8 mm, 5 µm column (Torrance, cat. 00H-2146-KO) with samples starting at pH 4.0 and pH 7.0. Samples at pH 7.0 were diluted in PBS; samples at pH 4.0 were generated by lowering the pH with 200 mM Citric Acid, pH 3.0. The flow rate was 0.5 ml/min for 30 min, isocratic. The ChemStation analog-to-digital converter was set to 25,000 units/mV, peak width 2 seconds, slit 4 nM (cat. 35900E). Radioactivity was detected with a raytest Ramona 90 in line with a standard Agilent 1100 HPLC module system.
Protein-G bead extraction and SDS-PAGE separation.
Triton X-100 (final concentration of 1% (v/v)) was added to fractions collected from the size-exclusion HPLC followed by the addition of Protein-G beads (GE Lifesciences Inc., cat. 17-0885-01). Samples were incubated overnight at 4°C with gentle rotation and then the beads were washed four times with PBS + 1% Triton X-100. Each sample was split and half of the sample was reduced using NuPage® Sample Reducing Agent (cat. NP0004). Beads were treated with NuPAGE® 4x LDS Sample Buffer (pH 8.4) (cat. NP0007) ± NuPAGE® Sample Reducing Agent and incubated at 99°C for 5 min; then applied to NuPAGE® 4–12% Bis-Tris gels (cat. NP0321BOX) with NuPAGE® 1x MOPS SDS Running buffer (cat. NP0001). Gels were stained with Coomassie Blue R250 dye. All NuPAGE® reagents were obtained from Invitrogen, Corp.
Mass spectrometric and bioinformatics analysis.
Samples excised from SDS-PAGE were treated as previously described in reference Citation38. Briefly, following rapid in-solution microwaveassisted tryptic digestion peptides were separated by reverse phase chromatography and eluted directly into a nanospray ionization source with a spray voltage of 2 kV and were analyzed using an LTQ XL-Orbitrap mass spectrometer (ThermoFisher). Precursor ions were analyzed in the FTMS at 60,000 resolution. MS/MS was performed in the LTQ with the instrument operated in data dependent mode whereby the top 10 most abundant ions were subjected for fragmentation. Data was searched using the Mascot Search Algorithm (Matrix Sciences) or by de novo interpretation.
For searching Mascot data: Search criteria included a full MS tolerance of 20 ppm, MS/MS tolerance of 0.5 Da with GlyGly on Lys, Oxidation of Methionine, +57 Da on Cys and Phosphorylation of ST and Y as variable modifications with up to 3 mis-cleavages. Data was searched against the Mammalian subset of the Swissprot database.
Financial Support
Genentech provided all funding for this research. D.B., A.W., A.E.R., B.L., J.P.S., L.D., B.Q.S. and M.S.D. hold a financial interest in Genentech. E.D. has no conflicts.
Abbreviations
| FGFR4 | = | fibroblast growth factor receptor 4 |
| chLD1 | = | a human chimera of LD1, a mouse monoclonal antibody against human FGFR4 |
| CDR | = | complementary determining region |
| C3 | = | complement component 3 |
| C3d | = | complement 3d |
| ko | = | knock-out |
| HCC | = | hepatocellular carcinoma |
| HPLC | = | high pressure liquid chromatography |
| ELISA | = | enzyme-linked immunosorbent assay |
| SDS-PAGE | = | sodium dodecyl sulfate polyacrylamide gel electrophoresis |
Figures and Tables
Figure 1 Variable domain sequences of mouse and humanized variants of anti-FGFR4. The amino acid sequences of mouse LD1 and humanized variants hLD1.vB and hLD1.v22 are aligned with the (A) human kappa I (huKI) and (B) human VH subgroup III (huIII) variable domain frameworks used in trastuzumab. Differences are highlighted in yellow and positions are numbered according to Kabat. Hypervariable regions that were grafted from mouse LD1 into the human variable Kappa I and subgroup III consensus frameworks were selected based on a combination of sequence, structural and contact CDR definitionsCitation16 and are boxed. Three vernier positions in the light chain were altered to restore affinity during humanization; these positions are not expected to be surface exposed.
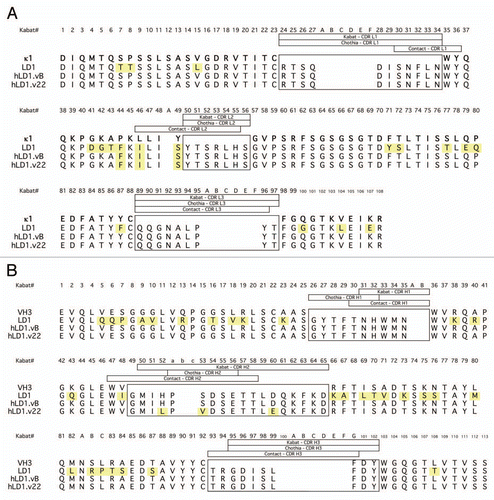
Figure 2 Pharmacokinetics and Distribution of anti-FGFR4 variants. (A) Comparison of the binding of chLD1 and hLD1.vB to FGFR4 using the FGFR4 ELISA. (B) Comparison of day 16 tumor volumes of chLD1, hLD1.vB and vehicle in an HUH7 human hepatocelluar carcinoma xenograft model in CRL nu/nu mice. Antibodies were administered at doses of 30 mg/kg twice weekly (10 mice per group). Only chLD1 was effective at reducing tumor growth relative to the PBS control (p value = 0.014) while hLD1.vB was not significantly effective (p value = 0.486). (C) Pharmacokinetics of chLD1 (open symbols) and hLD1.vB (closed symbols). NCR nude mice were IV administered 1 (dashed lines) or 20 (solid lines) mg/kg doses and samples were analyzed using the FGFR4 ELISA. Similar results were obtained using the IgG ELISA (not shown). (D) Tissue distribution of 125I-chLD1 and 125I-hLD1.vB in NCR nude mouse. Mice were administered either 125I-chLD1 (black bars) or 125I-hLD1.vB (open bars) and the percent of injected dose per gram of tissue (% ID/g) was determined at 2 h post-dose as described in Methods.
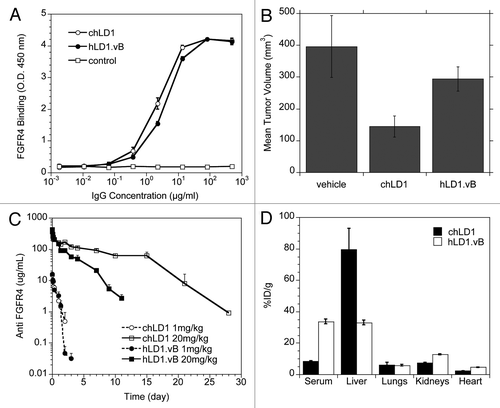
Figure 3 Identification of an interaction between hLD1.vB and mouse C3d. (A) Detection of chLD1 (black bar) and hLD1.vB (white bar) following incubation for 48 h in PBS/BSA or NCR nude mouse, rat, human and cynomolgus monkey plasma. Percent recovery was determined using the FGFR4 ELISA. (B) Plasma binding analysis of 125I-chLD1 (solid line) and 125I-hLD1.vB (dashed line). The traces are off-set; a dot indicates the position of the 150 kDa peaks. The antibodies were incubated in mouse plasma for 0 and 48 h followed by analysis using size exclusion HPLC. For incubations in PBS/BSA, human plasma or cynomolgus monkey plasma see Supplemental Figure 1. All incubations gave rise to a peak at the expected 150 kDa corresponding to IgG. Higher molecular weight peaks were only observed in mouse plasma samples containing hLD1.vB. The initial time point revealed additional peaks at ca. 270 and ca. 550 kDa while at 48 h, only the 270 kDa peak was observed. The high molecular weight peaks were not observed when hLD1.vB/mouse plasma incubations were performed at pH 4, indicating that the presence of these high molecular weight peaks was pH dependent (Sup. Fig. 1). (C) Immunoprecipitation of mouse plasma. chLD1 and hLD1.vB were incubated in mouse plasma for 24 h at 37°C and analyzed by size-exclusion HPLC. Protein-G beads were then added to fractions followed by SDS-PAGE analysis. A band at ca. 37 kDa was detected in fractions corresponding to the 270 kDa peak present in the hLD1.vB/mouse plasma sample (lane 3), but not in samples from cynomolgus monkey or human plasma or PBS/BSA (Sup. Fig. 1). This band was not observed in mouse plasma alone (lane 4) or mouse plasma incubated with chLD1 (lane 2). The protein molecular weight marker was run in lane 1. (D) MS/MS sequence coverage of mouse C3 obtained from immunoprecipitation with hLD1.vB. The sequence for mouse C3 is shown and identified peptides are highlighted in red; the region encoding C3d is underlined.
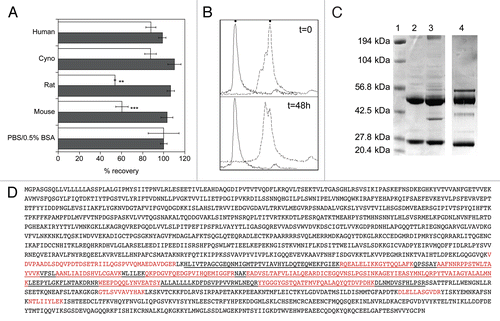
Figure 4 An affinity matured anti-FGFR4 variant lacks C3d binding. (A) Detection of FGFR4 binding of chLD1, hLD1.vB and hLD1.v22 incubated 16 h in NCR nude (black bar), C3 wt (open bar) or C3 ko (shaded bar) mouse serum prior to assessment using the FGFR4 ELISA. Samples were normalized to identical samples incubated in PBS/0.5% BSA. (B) Lack of mouse C3d immunoprecipitation by hLD1.v22. Immunoprecipitation from NCR nude mouse plasma using chLD1 (lane 2), hLD1.vB (lane 3) and hLD1.v22 (lane 4) was analyzed by SDS-PAGE. The band at ∼37 kDa was only detected in the hLD1.vB/mouse plasma sample. The protein molecular weight marker was run in lane 1.
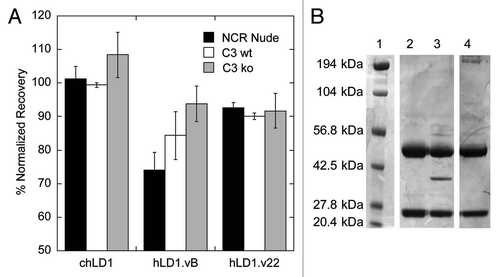
Figure 5 A loss in C3d binding restores Pharmacokinetics and Efficacy. (A) Pharmacokinetic analysis of chLD1 (open symbols) and hLD1.vB (solid symbols) in C3 wt (solid lines) and C3 ko (dashed lines) mice. Antibodies were IV administered at 20 mg/kg; their concentration in blood was monitored using the FGFR4 ELISA. The clearance of hLD1.vB in C3 ko mice is similar to that of chLD1 in both C3 ko and C3 wt mice. (B) Pharmacokinetic analysis of chLD1 (open circles), hLD1.vB (solid circles) and hLD1.v22 (solid diamond) in NCR nude mice. Antibodies were IV administered at 20 mg/kg; their concentration in blood was monitored using the FGFR4 ELISA. The clearance of hLD1.v22 is similar to that of chLD1. (C) Comparison of chLD1 (open circle), hLD1.vB (solid circle) and hLD1.v22 (solid diamond) in an HUH7 human HCC xenograft model in CRL nu/nu mice. Antibodies were administered at 30 mg/kg weekly (10 mice per group) and tumor volumes were monitored for 4 weeks. At day 21 chLD1 and hLD1.v22 (p value = 7 × 10−7 and 3 × 10−5, respectively) were effective at reducing tumor growth relative to the PBS control (open square) while hLD1.vB was only marginally effective (p value = 0.011).
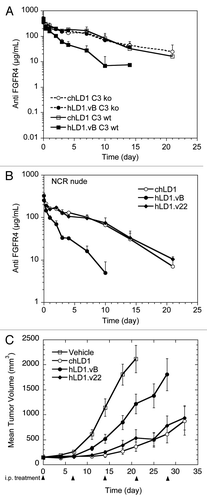
Table 1 Binding kinetics of anti-FGF.R4 variants
Additional material
Download Zip (467.2 KB)Acknowledgments
We gratefully acknowledge Misia Howell, Kirsten Messick, Michelle Schweiger and Shannon Stainton for conducting pharmacokinetic and biodistribution studies, Jennie Lill and Victoria Pham for MS sequencing, Greg Bennett for antibody labeling, Kenneth Katschke for the C3 ko and C3 wt mice, Phuong Tran and Blair Wilson for generic human Fc ELISA analysis, Sarajane Ross, Maria Hristopoulos, Rayna Venook, Kathy Parsons and Janet Tien for tumor efficacy studies, Kurt Schroeder for antibody purification and Menno van Lookeren Campagne for C3 related discussions.
References
- James LC, Roversi P, Tawfik DS. Antibody multispecificity mediated by conformational diversity. Science 2003; 299:1362 - 1367
- Krishnan L, Sahni G, Kaur KJ, Salunke DM. Role of antibody paratope conformational flexibility in the manifestation of molecular mimicry. Biophys J 2008; 94:1367 - 1376
- Notkins AL. Polyreactivity of antibody molecules. Trends Immunol 2004; 25:174 - 179
- Thorpe IF, Brooks CL 3rd. Molecular evolution of affinity and flexibility in the immune system. Proc Natl Acad Sci USA 2007; 104:8821 - 8826
- Yin J, Beuscher AE 4th, Andryski SE, Stevens RC, Schultz PG. Structural plasticity and the evolution of antibody affinity and specificity. J Mol Biol 2003; 330:651 - 656
- Thielges MC, Zimmermann J, Yu W, Oda M, Romesberg FE. Exploring the energy landscape of antibody-antigen complexes: protein dynamics, flexibility and molecular recognition. Biochemistry 2008; 47:7237 - 7247
- Feyen O, Lueking A, Kowald A, Stephan C, Meyer HE, Gobel U, et al. Off-target activity of TNFα inhibitors characterized by protein biochips. Analytical and Bioanalytical Chemistry 2008; 391:1713 - 1720
- Kijanka G, Ipcho S, Baars S, Chen H, Hadley K, Beveridge A, et al. Rapid characterization of binding specificity and cross-reactivity of antibodies using recombinant human protein arrays. J Immunol Methods 2009; 340:132 - 137
- Lueking A, Beator J, Patz E, Mullner S, Mehes G, Amersdorfer P. Determination and validation of off-target activities of anti-CD44 variant 6 antibodies using protein biochips and tissue microarrays. Biotechniques 2008; 45:1 - 5
- Wu H, Pfarr DS, Johnson S, Brewah YA, Woods RM, Patel NK, et al. Development of motavizumab, an ultra-potent antibody for the prevention of respiratory syncytial virus infection in the upper and lower respiratory tract. J Mol Biol 2007; 368:652 - 665
- Rosenwald S, Kafri R, Lancet D. Test of a statistical model for molecular recognition in biological repertoires. J Theor Biol 2002; 216:327 - 336
- James LC, Tawfik DS. The specificity of cross-reactivity: promiscuous antibody binding involves specific hydrogen bonds rather than nonspecific hydrophobic stickiness. Protein Sci 2003; 12:2183 - 2193
- Kramer A, Keitel T, Winkler K, Stocklein W, Hohne W, Schneider-Mergener J. Molecular basis for the binding promiscuity of an anti-p24 (HIV-1) monoclonal antibody. Cell 1997; 91:799 - 809
- French DM, Lin BC, Wang M, Hötzel K, Bolon B, Ferrando R, et al. Targeting FGFR4 Inhibits Hepatocellular Carcinoma in Preclinical Animal Models. PloS ONE 2011;
- Dennis MS. Shire S, Gombotz W, Bechtold-Peters K, Andya J. Humanization by CDR Repair. Pharmaceutical Aspects of Monoclonal Antibodies 2010; New York Co-published by the Association for Pharmaceutical Scientists & Springer 9 - 28
- MacCallum RM, Martin ACR, Thornton JT. Antibody-antigen interactions: Contact analysis and binding site topography. J Mol Biol 1996; 262:732 - 745
- Adams CW, Allison DE, Flagella K, Presta L, Clarke J, Dybdal N, et al. Humanization of a recombinant monoclonal antibody to produce a therapeutic HER dimerization inhibitor, pertuzumab. Cancer Immunol Immunother 2006; 55:717 - 727
- Kim SJ, Park Y, Hong HJ. Antibody engineering for the development of therapeutic antibodies. Mol Cells 2005; 20:17 - 29
- Lutz HU, Jelezarova E. Complement amplification revisited. Mol Immunol 2006; 43:2 - 12
- Sahu A, Lambris JD. Structure and biology of complement protein C3, a connecting link between innate and acquired immunity. Immunol Rev 2001; 180:35 - 48
- Manderson AP, Pickering MC, Botto M, Walport MJ, Parish CR. Continual low-level activation of the classical complement pathway. J Exp Med 2001; 194:747 - 756
- Thurman JM, Holers VM. The central role of the alternative complement pathway in human disease. J Immunol 2006; 176:1305 - 1310
- Sahu A, Pangburn MK. Covalent attachment of human complement C3 to IgG. J Biol Chem 1994; 269:28997 - 29002
- Vidarte L, Pastor C, Mas S, Blazquez AB, de los Rios V, Guerrero R, et al. Serine 132 is the C3 covalent attachment point on the CH1 domain of human IgG1. J Biol Chem 2001; 276:38217 - 38223
- Osmers I, Szalai AJ, Tenner AJ, Barnum SR. Complement in BuB/BnJ mice revisited: serum C3 levels and complement opsonic activity are not elevated. Mol Immunol 2006; 43:1722 - 1725
- Otte L, Knaute T, Schneider-Mergener J, Kramer A. Molecular basis for the binding polyspecificity of an anti-cholera toxin peptide 3 monoclonal antibody. J Mol Recognit 2006; 19:49 - 59
- Arenkov P, Kukhtin A, Gemmell A, Voloshchuck S, Chupeeva V, Mirzabekov A. Protein microchips: use for immunoassay and enzymatic reactions. Anal Biochem 2000; 278:123 - 131
- Ge H. UPA, a universal protein array system for quantitative detected protein-protein, protein-DNA, protein-RNA and protein-ligand interactions. Nucleic Acids Res 2000; 28:1 - 7
- Fodor SP, Read JL, Pirrung MC, Stryer L, Lu AT, Solas D. Light-directed, spatially addressable parallel chemical synthesis. Science 1991; 251:767 - 773
- Forster R, Valin M, Lemarchand T, Palate B, Fifre A. Validation of tissue cross reactivity (TCR) studies: Tissue cross reactivity of anti-human CD209 in cynomolgus tissues. Toxicol Lett 2009; 189:100
- Kallioniemi OP, Wagner U, Kononen J, Sauter G. Tissue microarray technology for high-throughput molecular profiling of cancer. Hum Mol Genet 2001; 10:657 - 662
- Lynch C, Grewel I. Chernajovsky Y, Nissim A. Preclinical safety evaluation of monoclonal antibodies. Therapeutic Antibodies: Handbook of Experimental Pharmacology 2008; Berlin Springer-Verlag 19 - 44
- Dennis MS. Shire SJ, Gombotz W, Bechtold-Peters K, Adndya J. CDR Repair: A novel approach to antibody humaization. Current Trends in Monoclonal Antibody Development and Manufacturing 2010; New York Springer 9 - 28
- Gallop MA, Barrett RW, Dower WJ, Fodor SPA, Gordon EM. Applications of combinatorial technologies to drug discovery. 1. Background and peptide combinatorial libraries. J Med Chem 1994; 37:1233 - 1251
- Lee CV, Liang WC, Dennis MS, Eigenbrot C, Sidhu SS, Fuh G. High-affinity human antibodies from phage-displayed synthetic Fab libraries with a single framework scaffold. J Mol Bio 2004; 340:1073 - 1093
- Circolo A, Garnier G, Fukuda W, Wang X, Hidvegi T, Szalai AJ, et al. Genetic disruption of the murine complement C3 promoter region generates deficient mice with extrahepatic expression of C3 mRNA. Immunopharmacology 1999; 42:135 - 149
- Chizzonite R, Truitt T, Podlaski FJ, Wolitzky AG, Quinn PM, Nunes P, et al. IL-12: Monocloncal antibodies specific for the 40 kDa subunit block receptor binding and biological activity on activated human lymphoblasts. J Immunol 1991; 147:1548 - 1556
- Jin Z, Li Y, Pitti R, Lawrence D, Pham VC, Lill JR, et al. Cullin3-based polyubiquitination and p62-dependent aggregation of caspase-8 mediate extrinsic apoptosis signaling. Cell 2009; 137:721 - 735