Abstract
Craniosynostosis describes the premature fusion of one or more cranial sutures and can lead to dramatic manifestations in terms of appearance and functional impairment. Contemporary approaches for this condition are primarily surgical and are associated with considerable morbidity and mortality. The additional post-operative problems of suture refusion and bony relapse may also necessitate repeated surgeries with their own attendant risks. Therefore, a need exists to not only optimize current strategies but also to develop novel biological therapies which could obviate the need for surgery and potentially treat or even prevent premature suture fusion. Clinical studies of patients with syndromic craniosynostosis have provided some useful insights into the important signaling pathways and molecular events guiding suture fate. Furthermore, the highly conserved nature of craniofacial development between humans and other species have permitted more focused and step-wise elucidation of the molecular underpinnings of craniosynostosis. This review will describe the clinical manifestations of craniosynostosis, reflect on our understanding of syndromic and non-syndromic craniosynostoses and outline the different approaches that have been adopted in our laboratory and elsewhere to better understand the pathogenesis of premature suture fusion. Finally, we will assess to what extent our improved understanding of the pathogenesis of craniosynostosis, achieved through laboratory-based and clinical studies, have made the possibility of a non-surgical pharmacological approach both realistic and tangible.
Keywords: :
Introduction
Craniosynostosis, a term first coined by Otto in 1830, describes the premature fusion of cranial sutures, the fibrous joints between calvarial bones.Citation1 Craniosynostosis can lead to dramatic clinical manifestations in terms of cosmesis and functional impairment and unsurprisingly therefore, literary and pictorial representations of this condition span history.Citation2,Citation3 Moreover, craniosynostosis has captivated the interest of both basic scientists and clinicians alike, by posing unique challenges to understanding its pathogenesis and to identifying appropriate and timely clinical approaches which are capable of providing safe, satisfactory, and sustainable long-term outcomes. Virchow, in the 1850s, was the first to state that following premature fusion of a calvarial suture, compensatory growth occurs in a plane parallel to the fused suture with minimal growth in a perpendicular plane.Citation4 Furthermore, he proposed that premature fusion was associated with inflammation of the meninges. In 1912, Crouzon made the key observation that there may be a genetic contribution to the pathogenesis of craniosynostosis, and in 1959, Moss proposed that craniosynostosis resulted from abnormal development of the cranial base leading to transmission of altered mechanical forces to the overlying suture.Citation5,Citation6 These accounts eventually resulted in a paradigm shift in clinical approach to craniosynostosis, with the acceptance that suturectomy alone was insufficient. Paul Tessier subsequently ushered in the modern era of craniofacial surgery with his description of total calvarial vault remodelling to increase intracranial volume and create a more normal appearance.Citation7,Citation8
While contemporary approaches to the treatment of craniosynostosis are based on these early pioneering reports, procedures used today still carry a mortality rate of 1.5–2% and are associated with considerable morbidity including blood loss, infection, CSF leak, and lengthy hospital stays as well as post-operative monitoring in an intensive care unit.Citation9-Citation11 Furthermore, additional post-operative problems include suture refusion and bony relapse necessitating repeated surgeries with their own attendant risks.Citation12,Citation13 Clearly, a need exists not only to optimize current strategies but to also develop novel biological therapies which may successfully treat or even prevent craniosynostosis, thereby obviating the need for surgery and its potentially deleterious consequences. In this review we will describe the clinical manifestations of craniosynostosis, reflect on our understanding of syndromic and non-syndromic craniosynostoses and outline the different approaches which have been adopted in our laboratory and elsewhere to better understand the pathogenesis of craniosynostosis. Finally, we will assess to what extent our improved understanding of the pathogenesis of craniosynostosis achieved through laboratory-based and clinical studies have made the possibility of a non-surgical pharmacological approach both realistic and tangible.
Suture Morphogenesis and Development of the Cranial Vault
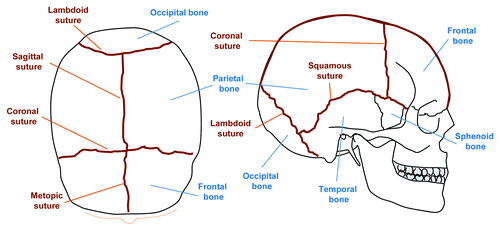
In order to understand the aberrations in suture biology which may lead to craniosynostosis, sutures should not simply be viewed as articulations between two bones.Citation2 Rather, they are sites of osteogenesis developed from embryonic mesenchyme. They constitute not only the advancing osteogenic fronts of the flat calvarial bones but also act as a niche for osteogenic progenitors which may proliferate or differentiate in a tightly regulated program orchestrated through appropriate molecular cues. The major sutures in humans are the metopic suture which separates the two frontal bones, the sagittal suture which separates the two parietal bones, the coronal suture where the parietal and frontal bones meet, and the lambdoid suture where parietal and occipital bones meet. While sutures form at the approximations of advancing fronts of the developing bones, what governs their location remains largely unknown.Citation14 Sutures not only allow for necessary deformation of the skull during passage though the birth canal but also subsequently act as important growth centers of the skull during early years of life.Citation3 The orchestration of this complex program is tightly controlled, and minor perturbations between developing tissues such as the brain, dura mater, osteogenic fronts, and suture mesenchyme may potentially lead to premature fusion of the sutures.
The membranous cranial vault consists of paired frontal and parietal bones as well as a portion of the occipital bone. These form through intramembranous ossification in which mesenchymal cells condense and differentiate into osteogenic cells without a prior chondrogenic anlagen.Citation2 Early studies provided conflicting evidence as to the embryonic origin of parietal and frontal bones with Noden and Le Lievre proposing a mixed origin hypothesis of neural crest and mesodermal tissues.Citation15,Citation16 In contrast, Couly et al. purported that the cranial vault was composed entirely of neural crest cells.Citation17 More recently, Jiang et al. were able to define the disparate origins of the calvarium by using a transgenic mouse with a Wnt-1-Cre construct and a R26R β-galactosidase reporter.Citation18 Employing this approach, the frontal bone was found to be of neural crest origin while the parietal bone was noted to be from paraxial mesoderm. The posterior frontal (PF) suture, analogous to the metopic suture in humans, is thus bounded by two neural crest-derived osteogenic fronts and the sagittal suture (SAG) is bounded by two paraxial mesoderm fronts. The coronal suture (COR) located between the frontal and parietal bones lies between osteogenic fronts of disparate embryonic origin. Importantly, while suture mesenchyme from PF, SAG, and coronal sutures all share a common neural crest origin, only the PF suture in mice and rats fuses in a predictable manner.Citation3,Citation19
Craniosynostosis: Clues from Clinical Genetics
Craniosynostosis is a heterogeneous condition which can involve the partial or complete premature fusion of one or more cranial sutures. With an incidence of 1:2,500 live births, it can present as part of a syndrome or more commonly as an isolated finding in non-syndromic craniosynostoses.Citation2,Citation14 Though each of the major sutures including the sagittal, coronal, metopic, and lambdoid sutures may be involved (), sagittal synostosis is the most common (40–55%), followed by coronal (20–25%), metopic (5–15%), multiple suture synostosis (5–15%) and lambdoid synostosis (0–5%).Citation20,Citation21 While a comprehensive description of the clinical phenotypes in syndromic and non-syndromic craniosynostoses is beyond the scope of this review, a brief outline of some of the expected deformities resulting from premature fusion of each of the individual sutures and some of the more notable syndromes will be highlighted.
Figure 1. Schematic representation of the major bones and sutures of the adult human cranium. Top view (left) and lateral view (right) of the calvarium, showing bones (blue) and sutures (brown). By the second year of life the metopic suture is normally closed.
Sagittal synostosis is the most common type of craniosynostosis and is most often seen in non-syndromic forms. Compensatory calvarial growth parallel to the suture can lead to an elongated, scaphocephalic skull, derived from the Greek word scaphos for boat and cephalos for head (). This “typical phenotype,” however, is not always seen and depends on the extent of sagittal suture which is affected. A palpable ridge and narrow skull are more common features.Citation21 Coronal synostosis can be unilateral or bilateral and is less common than sagittal synostosis. Unilateral synostosis leads to ipsilateral flattening of the forehead and compensatory contralateral frontal bossing. In addition, failure of the greater wing of the sphenoid to descend results in a “harlequin” deformity with elongation of the orbit superiorly and laterally. Bilateral coronal synostosis () leads to shortening in the anteroposterior direction and a brachycephalic deformity.Citation21 Metopic synostosis () is associated with trigonocephaly, or a triangular keel-shaped head. Widening and increased height in the parietal region can occur due to compensatory growth posteriorly.Citation21 Finally, lambdoid synostosis () is rare and often difficult to distinguish clinically from positional plagiocephaly. Premature fusion of the lambdoid suture leads to ipsilateral flattening of the occipital region with a compensatory mastoid bulge. Contralateral growth in the parietal region is also noted and the cranial base becomes tilted.
Figure 2. Sagittal synostosis (Scaphocepaly). Top view (A) and lateral view (B) schematics showing the direction of aberrant compensatory bone growth (red lines) following premature sagittal suture fusion leading to an elongated, scaphocephalic skull, derived from the Greek word scaphos for boat and cephalos for head.
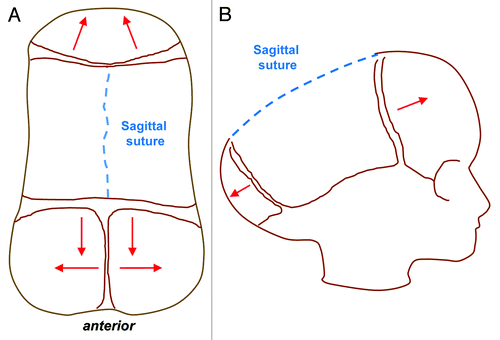
Figure 3. Bilateral coronal craniosynostosis. Top view (B) and lateral view (B) schematics showing the direction of compensatory calvarial growth (red lines) secondary to premature fusion of both coronal sutures (blue dashed lines) leading to shortening in the anteroposterior direction, increased height of the vault and therefore a turribrachycephalic deformity.
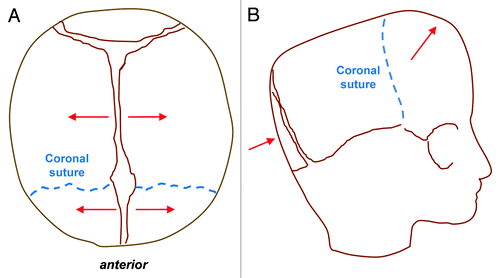
Figure 4. Metopic Synostosis. Top view (A) and lateral view (B) schematics demonstrating trigonocephaly, or a triangular keel-shaped head. Compensatory calvarial growth (red lines) can lead to widening and increased height in the parietal region due to compensatory growth in a posterior direction following premature fusion of the metopic suture (blue dashed line).
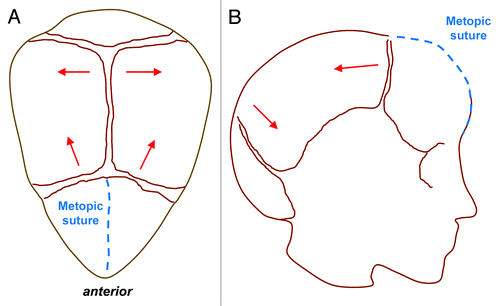
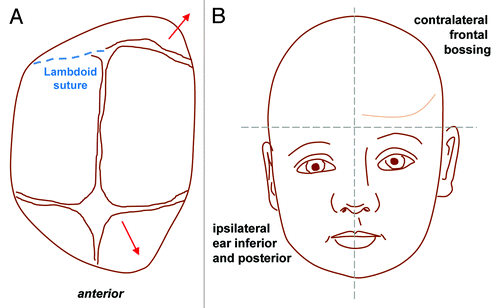
Figure 5. Lambdoid synostosis. Top view (A) and antero-posterior view (B) showing the direction of compensatory bone growth (red lines) following premature fusion of the lambdoid suture (blue dashed line). Lambdoid synostosis leads to ipsilateral flattening of the occipital region with a compensatory mastoid bulge which is a key distinguishing feature from positional (sometimes called positional or deformational) plagiocephaly from which it can be clinically difficult to differentiate. Contralateral growth in the parietal region is also noted and the cranial base becomes tilted.
While non-syndromic craniosynostoses are most frequently encountered, their genetic etiology remains poorly understood. EphrinA-4 (EFNA-4) is one of the first genes that when mutated has been associated with non-syndromic craniosynostosis.Citation22 Mutational analysis on cohorts of patients have also demonstrated a possible role for FGFRs-1, -2, -3 and TWIST-1.Citation23-Citation25 In contrast to non-syndromic craniosynostosis, genetic analysis of syndromic craniosynostosis has contributed a wealth of information, elucidating some of the important pathways for suture development and closure.Citation26 This has allowed for genetic screening and molecular diagnosis in patients with syndromic craniosynostosis which can be a useful adjunct to clinical diagnosis given the phenotypic heterogeneity. Importantly, it has also helped to direct studies on the molecular etiopathology of craniosynostosis. There are at least 150 syndromes associated with craniosynostosis as a major clinical feature. Linkage analysis in familial cases and molecular analysis of chromosomal alterations have identified several genes that when mutated are closely associated with syndromic craniosynostosis: FGFR-1, FGFR-2, FGFR-3, TWIST-1, EFNB-1, MSX-2 and more recently, RAB-23 ().Citation21
Table 1. Common mendelian causes of craniosynostosis
The FGFR family, which consists of four signal transduction receptor kinases, has drawn the most attention, as three of them have been associated with the majority of cases of syndromic craniosynostosis.Citation21 Upon dimerization, these receptors autophosphorylate and initiate signal transduction via a range of pathways to control key physiological processes including cell proliferation, differentiation, migration and apoptosis.Citation21,Citation27,Citation28 While a detailed account of the eponymous syndromes associated with FGFR mutations is beyond the scope of this review, it is worth highlighting some of the different biomolecular mechanisms, which can lead to premature suture fusion. Mutations in FGFRs-1, -2 and -3 are invariably gain of function mutations, primarily localized to the IgII - IgIII linker regions. This results in enhanced ligand affinity, more promiscuous ligand-receptor binding, or ligand-independent activation. Well documented mutations include the point substitution in FGFR-2 (Cys342Tyr) associated with Crouzon syndrome and Ser252Trp in Apert syndrome, FGFR-1 Pro252Arg in Pfeiffer syndrome, and analogous FGFR-3 mutations in Muenke syndrome.Citation29-Citation31 The prominent role played by FGF pathway gain of function mutations in syndromic craniosynostoses have prompted development of animal models and highlight potential targets for future biological intervention.
Transcription factors have also been implicated in the pathogenesis of craniosynostosis. TWIST-1, a basic helix-loop-helix transcription factor, is one such example that has drawn attention. Mutations in this gene are associated with Saethre-Chotzen syndrome, or acrocephalosyndactyly type III, an autosomal dominant condition characterized by unilateral or bilateral coronal synostosis.Citation32 TWIST-1 protein contains two highly conserved regions, a DNA binding domain and a basic helix-loop-helix motif. The majority of the mutations which have been described result in loss-of-function, leading to functional haploinsufficiency of TWIST-1.Citation33-Citation35 To date, over 100 distinct mutations in the TWIST-1 gene have been found to cause Saethre-Chotzen syndrome, including nucleotide substitutions (missense and nonsense), deletions, insertions, duplications and complex rearrangements.Citation36 Interestingly, genetic screening of patients with Saethre-Chotzen syndrome has also identified FGFR mutations, suggesting that TWIST-1 and FGFRs may be active in the same signaling network.Citation37 Experiments with human osteoblasts have highlighted a potential role for TWIST-1 to attenuate cellular response to FGF ligand, with overexpression of TWIST-1 leading to downregulation Early Growth Response Element-1, a known mediator of FGF signaling, and maintenance of an undifferentiated state.Citation38 In such a manner, TWIST-1 may thus contribute to continued suture patency.
MSX-2, a homeobox containing gene and potential modulator of downstream FGF activity, has also been associated with syndromic craniosynostosis.Citation39-Citation41MSX-2 mutation has been indentified in patients with Boston-type craniosynostosis, an autosomal dominant form of craniosynostosis.Citation32MSX-2 has been shown to be upregulated in response to increased FGF signaling and exogenous FGF-2 promotes MSX-2 expression and upregulation of markers for osteogenic differentiation.Citation39 Boston-type craniosynostosis involves a cytosine to adenosine base substitution resulting in the replacement of the amino acid proline with histidine (Pro7His). This amino acid change leads to premature suture ossification and other secondary deformities.Citation42
Ephrins, which are membrane-bound ligands for Eph family receptor tyrosine kinases, regulate cell adhesion and migration during development.Citation21 Mutations in EFNB-1 have been associated with craniofrontonasal syndrome, a developmental disorder that exhibits a very unusual pattern of X-linked inheritance with a paradoxically greater severity in heterozygous females than in hemizygous males.Citation26,Citation43,Citation44 Missense mutations constitute about 42% of EFNB-1 mutations.Citation45 The other major types of mutations include nonsense, frame-shift, and splice site, and lead to premature termination codons. Missense mutations change highly conserved amino acid residues in the extracellular ephrin domain and are expected to disrupt protein folding or interaction sites for EFNB-1 interacting partners resulting in loss-of-function. Craniofrontonasal syndrome is characterized by craniosynostosis, frontonasal dysplasia, orbital hypertelorism, a broad nasal tip, central nasal groove, and an anterior open bite.Citation21
While many of the genes so far identified with syndromic craniosynostosis have been shown to demonstrate autosomal dominant inheritance, RAB-23 mutations instead result in autosomal recessive transmission for craniosynostosis with Carpenter syndrome. Until 2001, only 40 cases of this condition had been reported.Citation21 Using homozygosity mapping, Jenkins et al.Citation46 identified five different mutations (four truncating and one missense) in RAB-23 which encodes a member of the RAB guanosine triphosphatase (GTPase) family of vesicle transport proteins involved in negatively regulating hedgehog singaling. Fusion of the midline (metopic and sagittal) sutures is typical in Carpenter syndrome and head shape can be variable. In severe cases, cloverleaf-shaped deformities have been described. Other well-recognized features include soft tissue syndactyly, short or missing middle phalanges of the hands and feet, and clinodactyly.
Lastly, though the role of TGF-β in bone biology has been well established, mutations in the TGF-β pathway, unlike the FGF signaling pathway, have not been closely associated with craniosynostosis. Hunter-Thompson Chondrodysplasia constituted the first clinical description of a TGF-β superfamily mutation in craniosynostosis.Citation47 Recently, an autosomal dominant gain of function mutation in TGF-β receptors has also been documented.Citation48 Despite the paucity of evidence for its role in clinical syndromes, both human and mouse studies have supported the importance of TGF-β signaling in suture morphogenesis and the maintenance of patency. Increased immunoreactivity for TGF-β2 was found on analysis of synostotic suture samples from 10 infants when compared with normal controls.Citation49 Murine studies have also documented upregulation of TGF-β2 within the PF suture during physiological fusion while TGF-β3 has been localized to the osteogenic fronts in patent sutures. A possible mechanism for these findings is that TGF-β3 may compete for receptors such as TGF-βR1 and in so doing, downregulate the pro-osteogenic effect of TGF-β2.Citation50 While clinical studies continue to define a role for TGF-β in the pathogenesis of craniosynostosis, available data nonetheless point to this pathway as a potentially important therapeutic target.
The Role of Animal Models in Cranial Suture Biology Research
With the description of clinical syndromes and an improved understanding of the genetics of craniosynostosis, some insights have been made into possible molecular events guiding suture fate. More in-depth definition of the complex pathways involved in premature pathologic suture fusion, however, has necessitated the development of animal models to study cranial suture development and craniosynostosis. These have included both mammalian models (e.g., mouse, rat and rabbit) and non-mammalian models (e.g., zebrafish and Xenopus laevis).
The murine model has proven to be an invaluable tool with which to study cranial sutures given the highly conserved nature of craniofacial development.Citation51,Citation52 The PF suture in both mice and rats undergo predictable physiological fusion in early postnatal life, affording investigators with the opportunity to study the process of normal suture fusion, while other sutures remain patent.Citation2 Our laboratory, among others, have chosen to study this elegant model to elucidate differences in the molecular signaling pathways and behavior of osteoblastic cells that differentiate between physiologic suture fusion and maintenance of suture patency. Differences in FGF signaling, TGF-β signaling, and Hedgehog signaling have been identified using the murine model.Citation53-Citation57 In addition, Warren et al. demonstrated that inhibitors of BMP signaling such as Noggin play a pivotal role in the differential fate of the murine PF suture. High FGF-2 activity in the PF suture was found to downregulate noggin expression at key time points thereby permitting unfettered endogenous BMP signaling and subsequent fusion of the suture.Citation58 More recently, Behr et al. showed that Wnt signaling is also likely to play a key role in suture fate.Citation59 Closure of the PF suture was found to be accompanied by a downregulation of canonical Wnt signaling, whereas patency of the SAG suture was associated with constitutively activated canonical Wnt signaling. Importantly, the fate of PF and SAG sutures could be reversed by manipulating Wnt signaling. Continuous activation of canonical Wnt signaling in the PF suture prevented endochondral ossification and suture closure whereas inhibition of canonical Wnt signaling in the SAG suture resulted in endochondral ossification and abnormal fusion.Citation59 Finally, the murine model has allowed for study of the stress induced system of intrauterine constraint. Originally developed by Koskinen-Moffett, Jacob et al. employed this system to show decreased expression of Indian hedgehog and noggin to be associated with constraint-induced suture fusion.Citation60,Citation61
Similar to mice and rats, a rabbit model of familial non-syndromic craniosynostosis has also been developed to study pathologic suture fusion.Citation62-Citation64 This model has demonstrated many similarities with the human condition: autosomal dominant transmission and variable phenotypic expression including unilaterally affected animals, delayed-onset suture synostosis and animals with complete bilateral fusion.Citation65 Moreover the size of the rabbit, in contrast to mice and rats, has facilitated studies looking at post-surgical re-stenosis. In rabbits with coronal synostosis, investigators have transplanted “wild-type” dura mater allografts into suturectomy sites following removal of pathologically fused sutures.Citation66 This resulted in a reduction of post-operative refusion rates, highlighting the important role dura mater plays in dictating overlying bone formation.
While significant advances in our understanding of cranial suture biology have been achieved using mammalian models, non-mammalian models possess some unique advantages which have rendered them invaluable. Our laboratory has used the zebrafish which possess a short reproductive cycle, large number of progeny, and a high degree of genetic and developmental conservation with humans.Citation67 Furthermore, they have transparent embryos which permits direct visualization of morphogenesis including that of the craniofacial elements without the need for much processing.Citation68 Genetic manipulation and screening on a whole genome basis can also be performed given that the entire genome has been sequenced. Interestingly, however, the zebrafish differs from humans and murine models in that the PF suture remains patent. Therefore, ongoing studies into the molecular mechanisms which confer this difference are being performed.
Over the past few years, our laboratory has also introduced Xenopus laevis as another potential model for studying suture biology.Citation69 In Xenopus, both the frontal and parietal bones are fused, leaving only two sutures, a midline suture that fuses during metamorphosis and a posterior suture that remains patent.Citation69 Xenopus laevis is also unique in that a dramatic transformation takes place from a cartilaginous to bony skull. A better understanding of the molecular program underpinning this process may provide novel insights into development of the calvarium.
The Role of Dura Mater
While investigations continue on defining the aberrant biomolecular events within the suture mesenchyme and osteogenic fronts, a paracrine effect of neural crest derived dura mater on overlying suture fate has also been purported from both in vivo and in vitro animal studies. Interruption of this interaction in the PF suture by interposition of a silicon sheet has been shown to delay suture fusion in rats.Citation70 Surgical relocation of the SAG suture over PF dura mater and PF suture over SAG dura mater resulted in abnormal SAG suture fusion and PF suture patency.Citation69,Citation71 Finally, transplantation of COR suture complexes without the underlying dura mater into parietal defects in rats revealed that in the absence of dura mater, the coronal suture fused. In contrast, when COR sutures were transplanted with the underlying associated dura mater, patency was maintained.Citation72
The importance of dura mater interaction with the overlying suture has also been highlighted by Mooney et al. using the rabbit model. Following coronal suturectomy, re-fusion could be prevented by dura mater allografts from WT rabbits allowing for normal unrestricted craniofacial growth.Citation66 More recently, in vitro co-culture experiments by Cooper and colleagues showed that associated dura mater from fused coronal sutures induced increased alkaline phosphatase activity in osteoblasts harvested from the fused coronal suture.Citation73 This effect was not observed, however, in osteoblasts derived from wild-type rabbits. Lastly, in vivo experiments similar to those performed in the rat have demonstrated that interposition of a physical barrier between the dura mater and coronal suture complex could successfully inhibit dura-mater-derived osteogenesis in rabbits.Citation74 Such invaluable studies have thus deepened our comprehension into the myriad of influences guiding suture fate and provide alternative avenues for subsequent development of therapeutics aimed at preventing post-operative resynostosis.
Future Directions: Approaching a New Era of Targeted Therapeutics
While the mainstay of treatment for craniosynostosis remains surgical, accumulating data from insightful clinical and animal studies have rendered the tantalising prospect of an entirely non-surgical approach to craniosynostosis possible. Furthermore, advances in our understanding of biomolecular mechanisms involved in suture fusion along with an evolution of surgical approaches may permit the use of combination therapies which would employ improved surgical technique in concert with pharmacological or genetic adjuncts to minimize morbidities of total cranial vault remodelling.
The first reported surgical intervention for sagittal synostosis was made by Lannelongue in 1890 who described a strip craniectomy to remove the prematurely fused suture. Early attempts at surgical correction, however, were plagued by post-operative refusion at craniectomy sites. Alternative techniques have subsequently been described including the pioneering work of Paul Tessier in the 1970s who developed an approach for total calvarial vault remodeling.Citation7 This allowed for immediate volumetric expansion and correction of the misshapen skull but has also been associated with increased operative times, hospital stays, and blood loss requiring transfusion.Citation8-Citation12 In 1998, Jimenez and Barone described the first minimally invasive endoscopic repair for sagittal synostosis, demonstrating shorter operative times, reduced blood loss, and earlier hospital discharge.Citation75 Importantly, however, minimally invasive approaches achieve less immediate remodeling and instead rely on the adjunctive post-operative use of skull-molding helmets which can range from several months to a year. Nevertheless, endoscopic cranial suture release is frequently performed and is the favored technique at many centers for certain types of craniosynostosis.Citation11
Although the evolution of surgical technique is a vital aspect of improving clinical care and outcome for patients with craniosynostosis, the significant advances in our understanding of the biomolecular underpinnings guiding cranial suture development hold promise for a potential paradigm shift in treatment and/or prevention of craniosynostosis. Given the well-established role of FGF signaling in the etiopathogenesis of craniosynostosis, FGF receptor downregulation or interference with the signaling cascade presents an avenue for development of pharmacological therapy. Ueno et al.Citation76 first described use of a truncated FGFR-1, which lacks its cytoplasmic domain, to inhibit signal transduction by FGF ligands.Citation76 Greenwald et al. subsequently employed the same truncated FGFR-1 in rat calvarial osteoblasts, demonstrating impaired MAP kinase activation in response to FGF-253. Furthermore, when this dominant-negative FGF receptor was introduced in utero into the PF sutures of fetal rats, postnatal fusion of the PF suture was prevented. This elegant study demonstrated the possibility that by reducing the physiological level of FGF signaling through use of a truncated receptor, abnormal suture fusion may be prevented. Alternatively, RNA interference could be used to target suppression of specific FGF receptors in patients with gain-of-function mutations. Promising results have been demonstrated in mice using a shRNA targeting a mutant form of FGFR-2.Citation77 This was shown to prevent an Apert-like syndrome in mice. Importantly, though, while this technique could provide a highly specific and efficient reduction in gene expression, clinical translation would demand the need for development of a means for safe, targeted delivery and durable suppression throughout calvarial development.
Modulation of downstream FGF signal transduction is another appealing target for directed pharmacological therapy. Treatment of Apert syndrome osteoblasts with the MAP kinase inhibitor PD98059 has been shown to reduce IL-1α expression which is pathologically upregulated in these cells.Citation78 Similar inhibitors of the MAPK pathway may also impede pathological suture fusion resulting from increased FGF signaling activity.Citation79 In a mouse model of Apert syndrome, treatment of mutant mice with U0126 significantly inhibited craniosynsotosis.Citation77 These findings highlight the potential for small-molecule inhibitors to be used in the treatment of specific gain-of-function mutations observed in syndromic craniosynostosis.
The TGFβ signaling pathway has also attracted significant attention as a potential target for pharmacological therapy. Given elevated isoforms of TGF-β in prematurely fused sutures, Opperman et al. demonstrated that delivery of TGF-β antibodies could alter suture fate in an ex vivo murine organ culture system.Citation50,Citation80,Citation81 Delivery of neutralizing antibodies to TGF-β3 resulted in aberrant suture fusion while TGF-β2 antibodies was found to rescue sutures from osseous obliteration.Citation81 More recently, suppression of TGF-β1 through RNA interference in mouse primary dura cell culture has been shown to downregulate expression of FGF-2 and FGFR-1.Citation82,Citation83 This again raises the exciting prospect of using siRNAs to suppress genes involved in promoting suture fusion. But as mentioned above, an efficient and safe means to deliver siRNA constructs must be first developed to achieve clinical efficacy and ongoing studies are evaluating a variety of synthetic vehicles including nanoparticles, liposomes, and other lipid-like materials.Citation84
While investigations continue to develop novel pharmacological approaches targeting the biomolecular pathways involved in pathologic suture fusion, studies have also identified potential candidates that may serve as useful adjuncts to contemporary surgical treatment. As previously described, Warren et al. demonstrated that overexpression of the BMP antagonist Noggin could prevent physiologic suture fusion in mice.Citation58 In contrast, studies have also shown downregulation of Noggin to enhance signaling by endogenous BMPs resulting in increased osteogenesis. Based on these findings, Cooper and colleagues have evaluated the ability for exogenous delivery of Noggin to inhibit postoperative resynostosis in both mice and rabbits.Citation85,Citation86 Introduction of Noggin in a slow-resorbing collagen vehicle was found to significantly improve maintenance of post-suturectomy defects compared with control animals which demonstrated complete re-ossification. These data thus suggest that Noggin may have a potential therapeutic role as an adjunct to surgery, preventing postoperative re-fusion in children with craniosynostosis.
Conclusion
As our understanding of the molecular and genetic underpinnings of craniosynostosis improves, the prospect of pharmacological or genetic therapy for the treatment or prevention of premature suture fusion becomes increasingly tangible. Such an approach, however, has yet to overcome significant obstacles in order to translate into the clinical setting. The demonstration of multiple signaling pathways involved in guiding suture fate and the complexity of cross talk between these pathways may render a one pathway pharmacological approach insufficient. Moreover, the highly conserved pathways which have been associated with craniosynostosis also play a variety of key physiological roles during development. Therefore, any potential therapy must be focused in a spatiotemporal manner to avoid unwanted deleterious consequences. Contemporary approaches to this heterogeneous and potentially debilitating condition remain surgical, and evolution of current techniques continues with the goal of minimizing morbidities associated with such surgical interventions. Toward this end, exploitation of targeted pharmacological therapy as an adjunct to prevent post-operative suture re-fusion may be readily incorporated. Nevertheless, the promise for novel pharmacological/genetic-based therapies has become increasingly real, and the possibility to obviate need for surgery remains an enticing prospect. With continued studies in clinical genetics and biomolecular mechanisms involved in premature suture fusion, new insights will undoubtedly be made which could, the in the future, usher in a new era of non-surgical therapy for craniosynostosis.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Otto. A. Lehrbuch der Pathologischen Anatomie. Berlin, Germany: Rucher., 1830.
- Lenton KA, Nacamuli RP, Wan DC, Helms JA, Longaker MT. Cranial suture biology. Curr Top Dev Biol 2005; 66:287 - 328; http://dx.doi.org/10.1016/S0070-2153(05)66009-7; PMID: 15797457
- Slater BJ, Lenton KA, Kwan MD, Gupta DM, Wan DC, Longaker MT. Cranial sutures: a brief review. Plast Reconstr Surg 2008; 121:170e - 8e; http://dx.doi.org/10.1097/01.prs.0000304441.99483.97; PMID: 18349596
- Virchow R. Berh Phyd Med Gesellsch Wuerzburg. 1851; 2:231 - 71
- Moss ML. The pathogenesis of premature cranial synostosis in man. Acta Anat (Basel) 1959; 37:351 - 70; http://dx.doi.org/10.1159/000141479; PMID: 14424622
- Crouzon O. Dysostose cranio-faciale hereditaire. Bull Mem Soc Med Hop Paris 1912; 33:545 - 55
- Tessier P. [Total facial osteotomy. Crouzon’s syndrome, Apert’s syndrome: oxycephaly, scaphocephaly, turricephaly]. Ann Chir Plast 1967; 12:273 - 86; PMID: 5622570
- Tessier P. [The treatment of facial dysmorphy peculiary to cranio-facial dysostosis (C.F.D.). Crouzon and Apert diseases. Total osteotomy and sagittal displacement of the facial mass]. Chirurgie 1970; 96:667 - 74; PMID: 5450453
- Grabb WC, Smith JW, Aston SJ. Plastic Surgery, 4th Edition. Boston: Little Brown, 1991.
- Whitaker LA, Munro IR, Salyer KE, Jackson IT, Ortiz-Monasterio F, Marchac D. Combined report of problems and complications in 793 craniofacial operations. Plast Reconstr Surg 1979; 64:198 - 203; http://dx.doi.org/10.1097/00006534-197908000-00011; PMID: 377338
- Proctor MR. Endoscopic cranial suture release for the treatment of craniosynostosis--is it the future?. J Craniofac Surg 2012; 23:225 - 8; http://dx.doi.org/10.1097/SCS.0b013e318241b8f6; PMID: 22337414
- Marchac D, Renier D. New aspects of craniofacial surgery. World J Surg 1990; 14:725 - 32; http://dx.doi.org/10.1007/BF01670519; PMID: 2256344
- McCarthy JG, Epstein F, Sadove M, Grayson B, Zide B. Early surgery for craniofacial synostosis: an 8-year experience. Plast Reconstr Surg 1984; 73:521 - 33; http://dx.doi.org/10.1097/00006534-198404000-00001; PMID: 6709733
- Wilkie AO, Morriss-Kay GM. Genetics of craniofacial development and malformation. Nat Rev Genet 2001; 2:458 - 68; http://dx.doi.org/10.1038/35076601; PMID: 11389462
- Noden DM. Interactions and fates of avian craniofacial mesenchyme. Development 1988; 103:Suppl 121 - 40; PMID: 3074905
- Le Lièvre CS. Participation of neural crest-derived cells in the genesis of the skull in birds. J Embryol Exp Morphol 1978; 47:17 - 37; PMID: 722230
- Couly GF, Coltey PM, Le Douarin NM. The triple origin of skull in higher vertebrates: a study in quail-chick chimeras. Development 1993; 117:409 - 29; PMID: 8330517
- Jiang X, Iseki S, Maxson RE, Sucov HM, Morriss-Kay GM. Tissue origins and interactions in the mammalian skull vault. Dev Biol 2002; 241:106 - 16; http://dx.doi.org/10.1006/dbio.2001.0487; PMID: 11784098
- Morriss-Kay GM, Wilkie AO. Growth of the normal skull vault and its alteration in craniosynostosis: insights from human genetics and experimental studies. J Anat 2005; 207:637 - 53; http://dx.doi.org/10.1111/j.1469-7580.2005.00475.x; PMID: 16313397
- Cohen MM Jr., Craniosynostosis MR. Diagnosis, Evaluation and Management., 2nd Edition. Ed. New York.: Oxford University Press., 2000.
- Rice DP. Craniofacial sutures. Development, disease and treatment. Preface. Front Oral Biol 2008; 12:xi; PMID: 18491429
- Merrill AE, Bochukova EG, Brugger SM, Ishii M, Pilz DT, Wall SA, et al. Cell mixing at a neural crest-mesoderm boundary and deficient ephrin-Eph signaling in the pathogenesis of craniosynostosis. Hum Mol Genet 2006; 15:1319 - 28; http://dx.doi.org/10.1093/hmg/ddl052; PMID: 16540516
- Lattanzi W, Bukvic N, Barba M, Tamburrini G, Bernardini C, Michetti F, et al. Genetic basis of single-suture synostoses: genes, chromosomes and clinical implications. Childs Nerv Syst 2012; 28:1301 - 10; http://dx.doi.org/10.1007/s00381-012-1781-1; PMID: 22872241
- Mefford HC, Shafer N, Antonacci F, Tsai JM, Park SS, Hing AV, et al. Copy number variation analysis in single-suture craniosynostosis: multiple rare variants including RUNX2 duplication in two cousins with metopic craniosynostosis. Am J Med Genet A 2010; 152A:2203 - 10; http://dx.doi.org/10.1002/ajmg.a.33557; PMID: 20683987
- Cunningham ML, Horst JA, Rieder MJ, Hing AV, Stanaway IB, Park SS, et al. IGF1R variants associated with isolated single suture craniosynostosis. Am J Med Genet A 2011; 155A:91 - 7; http://dx.doi.org/10.1002/ajmg.a.33781; PMID: 21204214
- Passos-Bueno MR, Serti Eacute AE, Jehee FS, Fanganiello R, Yeh E. Genetics of craniosynostosis: genes, syndromes, mutations and genotype-phenotype correlations. Front Oral Biol 2008; 12:107 - 43; http://dx.doi.org/10.1159/000115035; PMID: 18391498
- Givol DEV, Lonai P. The fibroblast growth factor signalling pathway. In: Epstein CJ ER, Wynshaw-Boris A., ed. Inborn errors of development. Oxford.: Oxford University Press., 2004.:367-79.
- Eswarakumar VP, Lax I, Schlessinger J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev 2005; 16:139 - 49; http://dx.doi.org/10.1016/j.cytogfr.2005.01.001; PMID: 15863030
- Wilkie AO, Slaney SF, Oldridge M, Poole MD, Ashworth GJ, Hockley AD, et al. Apert syndrome results from localized mutations of FGFR2 and is allelic with Crouzon syndrome. Nat Genet 1995; 9:165 - 72; http://dx.doi.org/10.1038/ng0295-165; PMID: 7719344
- Robin NH, Feldman GJ, Mitchell HF, Lorenz P, Wilroy RS, Zackai EH, et al. Linkage of Pfeiffer syndrome to chromosome 8 centromere and evidence for genetic heterogeneity. Hum Mol Genet 1994; 3:2153 - 8; http://dx.doi.org/10.1093/hmg/3.12.2153; PMID: 7881412
- Muenke M, Gripp KW, McDonald-McGinn DM, Gaudenz K, Whitaker LA, Bartlett SP, et al. A unique point mutation in the fibroblast growth factor receptor 3 gene (FGFR3) defines a new craniosynostosis syndrome. Am J Hum Genet 1997; 60:555 - 64; PMID: 9042914
- Patel A, Terner J, Travieso R, Clune JE, Steinbacher D, Persing JA. On Bernard Sarnat’s 100th birthday: pathology and management of craniosynostosis. J Craniofac Surg 2012; 23:105 - 12; http://dx.doi.org/10.1097/SCS.0b013e318240fb0d; PMID: 22337384
- Reid CS, McMorrow LE, McDonald-McGinn DM, Grace KJ, Ramos FJ, Zackai EH, et al. Saethre-Chotzen syndrome with familial translocation at chromosome 7p22. Am J Med Genet 1993; 47:637 - 9; http://dx.doi.org/10.1002/ajmg.1320470511; PMID: 8266989
- Wilkie AO, Yang SP, Summers D, Poole MD, Reardon W, Winter RM. Saethre-Chotzen syndrome associated with balanced translocations involving 7p21: three further families. J Med Genet 1995; 32:174 - 80; http://dx.doi.org/10.1136/jmg.32.3.174; PMID: 7783164
- Gripp KW, Zackai EH, Stolle CA. Mutations in the human TWIST gene. Hum Mutat 2000; 15:479; http://dx.doi.org/10.1002/(SICI)1098-1004(200005)15:5<479::AID-HUMU11>3.0.CO;2-X; PMID: 10790211
- Cunningham ML, Seto ML, Ratisoontorn C, Heike CL, Hing AV. Syndromic craniosynostosis: from history to hydrogen bonds. Orthod Craniofac Res 2007; 10:67 - 81; http://dx.doi.org/10.1111/j.1601-6343.2007.00389.x; PMID: 17552943
- Jabs EW. A TWIST in the fate of human osteoblasts identifies signaling molecules involved in skull development. J Clin Invest 2001; 107:1075 - 7; http://dx.doi.org/10.1172/JCI12853; PMID: 11342569
- Lee MS, Lowe GN, Strong DD, Wergedal JE, Glackin CA. TWIST, a basic helix-loop-helix transcription factor, can regulate the human osteogenic lineage. J Cell Biochem 1999; 75:566 - 77; http://dx.doi.org/10.1002/(SICI)1097-4644(19991215)75:4<566::AID-JCB3>3.0.CO;2-0; PMID: 10572240
- Ignelzi MA Jr., Wang W, Young AT. Fibroblast growth factors lead to increased Msx2 expression and fusion in calvarial sutures. J Bone Miner Res 2003; 18:751 - 9; http://dx.doi.org/10.1359/jbmr.2003.18.4.751; PMID: 12674336
- Liu YH, Tang Z, Kundu RK, Wu L, Luo W, Zhu D, et al. Msx2 gene dosage influences the number of proliferative osteogenic cells in growth centers of the developing murine skull: a possible mechanism for MSX2-mediated craniosynostosis in humans. Dev Biol 1999; 205:260 - 74; http://dx.doi.org/10.1006/dbio.1998.9114; PMID: 9917362
- Liu YH, Kundu R, Wu L, Luo W, Ignelzi MA Jr., Snead ML, et al. Premature suture closure and ectopic cranial bone in mice expressing Msx2 transgenes in the developing skull. Proc Natl Acad Sci U S A 1995; 92:6137 - 41; http://dx.doi.org/10.1073/pnas.92.13.6137; PMID: 7597092
- Müller U, Steinberger D, Kunze S. Molecular genetics of craniosynostotic syndromes. Graefes Arch Clin Exp Ophthalmol 1997; 235:545 - 50; http://dx.doi.org/10.1007/BF00947081; PMID: 9342602
- Twigg SR, Kan R, Babbs C, Bochukova EG, Robertson SP, Wall SA, et al. Mutations of ephrin-B1 (EFNB1), a marker of tissue boundary formation, cause craniofrontonasal syndrome. Proc Natl Acad Sci U S A 2004; 101:8652 - 7; http://dx.doi.org/10.1073/pnas.0402819101; PMID: 15166289
- Wieland I, Jakubiczka S, Muschke P, Cohen M, Thiele H, Gerlach KL, et al. Mutations of the ephrin-B1 gene cause craniofrontonasal syndrome. Am J Hum Genet 2004; 74:1209 - 15; http://dx.doi.org/10.1086/421532; PMID: 15124102
- Zafeiriou DI, Pavlidou EL, Vargìami E. Diverse clinical and genetic aspects of craniofrontonasal syndrome. Pediatr Neurol 2011; 44:83 - 7; http://dx.doi.org/10.1016/j.pediatrneurol.2010.10.012; PMID: 21215906
- Jenkins D, Baynam G, De Catte L, Elcioglu N, Gabbett MT, Hudgins L, et al. Carpenter syndrome: extended RAB23 mutation spectrum and analysis of nonsense-mediated mRNA decay. Hum Mutat 2011; 32:E2069 - 78; http://dx.doi.org/10.1002/humu.21457; PMID: 21412941
- Thomas JT, Lin K, Nandedkar M, Camargo M, Cervenka J, Luyten FP. A human chondrodysplasia due to a mutation in a TGF-beta superfamily member. Nat Genet 1996; 12:315 - 7; http://dx.doi.org/10.1038/ng0396-315; PMID: 8589725
- Loeys BL, Chen J, Neptune ER, Judge DP, Podowski M, Holm T, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet 2005; 37:275 - 81; http://dx.doi.org/10.1038/ng1511; PMID: 15731757
- Roth DA, Gold LI, Han VK, McCarthy JG, Sung JJ, Wisoff JH, et al. Immunolocalization of transforming growth factor beta 1, beta 2, and beta 3 and insulin-like growth factor I in premature cranial suture fusion. Plast Reconstr Surg 1997; 99:300 - 9, discussion 310-6; http://dx.doi.org/10.1097/00006534-199702000-00002; PMID: 9030135
- Opperman LA, Galanis V, Williams AR, Adab K. Transforming growth factor-beta3 (Tgf-beta3) down-regulates Tgf-beta3 receptor type I (Tbetar-I) during rescue of cranial sutures from osseous obliteration. Orthod Craniofac Res 2002; 5:5 - 16; http://dx.doi.org/10.1034/j.1600-0544.2002.01179.x; PMID: 12071374
- Opperman LA. Cranial sutures as intramembranous bone growth sites. Dev Dyn 2000; 219:472 - 85; http://dx.doi.org/10.1002/1097-0177(2000)9999:9999<::AID-DVDY1073>3.0.CO;2-F; PMID: 11084647
- Sahar DE, Longaker MT, Quarto N. Sox9 neural crest determinant gene controls patterning and closure of the posterior frontal cranial suture. Dev Biol 2005; 280:344 - 61; http://dx.doi.org/10.1016/j.ydbio.2005.01.022; PMID: 15882577
- Greenwald JA, Mehrara BJ, Spector JA, Warren SM, Fagenholz PJ, Smith LE, et al. In vivo modulation of FGF biological activity alters cranial suture fate. Am J Pathol 2001; 158:441 - 52; http://dx.doi.org/10.1016/S0002-9440(10)63987-9; PMID: 11159182
- James AW, Xu Y, Wang R, Longaker MT. Proliferation, osteogenic differentiation, and fgf-2 modulation of posterofrontal/sagittal suture-derived mesenchymal cells in vitro. Plast Reconstr Surg 2008; 122:53 - 63; http://dx.doi.org/10.1097/PRS.0b013e31817747b5; PMID: 18594386
- Mehrara BJ, Spector JA, Greenwald JA, Ueno H, Longaker MT. Adenovirus-mediated transmission of a dominant negative transforming growth factor-beta receptor inhibits in vitro mouse cranial suture fusion. Plast Reconstr Surg 2002; 110:506 - 14; http://dx.doi.org/10.1097/00006534-200208000-00022; PMID: 12142669
- James AW, Xu Y, Lee JK, Wang R, Longaker MT. Differential effects of TGF-beta1 and TGF-beta3 on chondrogenesis in posterofrontal cranial suture-derived mesenchymal cells in vitro. Plast Reconstr Surg 2009; 123:31 - 43; http://dx.doi.org/10.1097/PRS.0b013e3181904c19; PMID: 19116522
- Kim HJ, Rice DP, Kettunen PJ, Thesleff I. FGF-, BMP- and Shh-mediated signalling pathways in the regulation of cranial suture morphogenesis and calvarial bone development. Development 1998; 125:1241 - 51; PMID: 9477322
- Warren SM, Brunet LJ, Harland RM, Economides AN, Longaker MT. The BMP antagonist noggin regulates cranial suture fusion. Nature 2003; 422:625 - 9; http://dx.doi.org/10.1038/nature01545; PMID: 12687003
- Behr B, Longaker MT, Quarto N. Differential activation of canonical Wnt signaling determines cranial sutures fate: a novel mechanism for sagittal suture craniosynostosis. Dev Biol 2010; 344:922 - 40; http://dx.doi.org/10.1016/j.ydbio.2010.06.009; PMID: 20547147
- Jacob S, Wu C, Freeman TA, Koyama E, Kirschner RE. Expression of Indian Hedgehog, BMP-4 and Noggin in craniosynostosis induced by fetal constraint. Ann Plast Surg 2007; 58:215 - 21; http://dx.doi.org/10.1097/01.sap.0000232833.41739.a5; PMID: 17245153
- Persing JEM, James HE. Scientific foundations and surgical treatment of craniosynostosis. In: Moffett. LK-MaB, ed. Sutures and intruterine deformation. Baltimore.: Williams and Wilkins., 1989.:96-106.
- Mooney MP, Losken HW, Tschakaloff A, Siegel MI, Losken A, Lalikos JF. Congenital bilateral coronal suture synostosis in a rabbit and craniofacial growth comparisons with experimental models. Cleft Palate Craniofac J 1993; 30:121 - 8; http://dx.doi.org/10.1597/1545-1569(1993)030<0121:CBCSSI>2.3.CO;2; PMID: 8452830
- Mooney MP, Losken HW, Siegel MI, Lalikos JF, Losken A, Smith TD, et al. Development of a strain of rabbits with congenital simple nonsyndromic coronal suture synostosis. Part I: Breeding demographics, inheritance pattern, and craniofacial anomalies. Cleft Palate Craniofac J 1994; 31:1 - 7; http://dx.doi.org/10.1597/1545-1569(1994)031<0001:DOASOR>2.3.CO;2; PMID: 8130237
- Mooney MP, Siegel MI, Burrows AM, Smith TD, Losken HW, Dechant J, et al. A rabbit model of human familial, nonsyndromic unicoronal suture synostosis. II. Intracranial contents, intracranial volume, and intracranial pressure. Childs Nerv Syst 1998; 14:247 - 55; http://dx.doi.org/10.1007/s003810050220; PMID: 9694336
- Mooney MP, Aston CE, Siegel MI, Losken HW, Smith TD, Burrows AM, et al. Craniosynostosis with autosomal dominant transmission in New Zealand white rabbits. J Craniofac Genet Dev Biol 1996; 16:52 - 63; PMID: 8675615
- Mooney MP, Burrows AM, Smith TD, Losken HW, Opperman LA, Dechant J, et al. Correction of coronal suture synostosis using suture and dura mater allografts in rabbits with familial craniosynostosis. Cleft Palate Craniofac J 2001; 38:206 - 25; http://dx.doi.org/10.1597/1545-1569(2001)038<0206:COCSSU>2.0.CO;2; PMID: 11386428
- Sheehan-Rooney K, Pálinkášová B, Eberhart JK, Dixon MJ. A cross-species analysis of Satb2 expression suggests deep conservation across vertebrate lineages. Dev Dyn 2010; 239:3481 - 91; http://dx.doi.org/10.1002/dvdy.22483; PMID: 21089028
- Quarto N, Longaker MT. The zebrafish (Danio rerio): a model system for cranial suture patterning. Cells Tissues Organs 2005; 181:109 - 18; http://dx.doi.org/10.1159/000091100; PMID: 16534205
- Slater BJ, Liu KJ, Kwan MD, Quarto N, Longaker MT. Cranial osteogenesis and suture morphology in Xenopus laevis: a unique model system for studying craniofacial development. PLoS One 2009; 4:e3914; http://dx.doi.org/10.1371/journal.pone.0003914; PMID: 19156194
- Slater BJ, Kwan MD, Gupta DM, Lee JK, Longaker MT. The role of regional posterior frontal dura mater in the overlying suture morphology. Plast Reconstr Surg 2009; 123:463 - 9; http://dx.doi.org/10.1097/PRS.0b013e3181954d21; PMID: 19182602
- Roth DA, Bradley JP, Levine JP, McMullen HF, McCarthy JG, Longaker MT. Studies in cranial suture biology: part II. Role of the dura in cranial suture fusion. Plast Reconstr Surg 1996; 97:693 - 9; http://dx.doi.org/10.1097/00006534-199604000-00001; PMID: 8628762
- Bradley JP, Levine JP, McCarthy JG, Longaker MT. Studies in cranial suture biology: regional dura mater determines in vitro cranial suture fusion. Plast Reconstr Surg 1997; 100:1091 - 9, 1100-2; http://dx.doi.org/10.1097/00006534-199710000-00001; PMID: 9326769
- Opperman LA, Sweeney TM, Redmon J, Persing JA, Ogle RC. Tissue interactions with underlying dura mater inhibit osseous obliteration of developing cranial sutures. Dev Dyn 1993; 198:312 - 22; http://dx.doi.org/10.1002/aja.1001980408; PMID: 8130378
- Cooper GM, Durham EL, Cray JJ Jr., Siegel MI, Losee JE, Mooney MP. Tissue interactions between craniosynostotic dura mater and bone. J Craniofac Surg 2012; 23:919 - 24; http://dx.doi.org/10.1097/SCS.0b013e31824e645f; PMID: 22627405
- Barone CM, Jimenez DF. Endoscopic craniectomy for early correction of craniosynostosis. Plast Reconstr Surg 1999; 104:1965 - 73, discussion 1974-5; http://dx.doi.org/10.1097/00006534-199912000-00003; PMID: 11149758
- Ueno H, Gunn M, Dell K, Tseng A Jr., Williams L. A truncated form of fibroblast growth factor receptor 1 inhibits signal transduction by multiple types of fibroblast growth factor receptor. J Biol Chem 1992; 267:1470 - 6; PMID: 1309784
- Shukla V, Coumoul X, Wang RH, Kim HS, Deng CX. RNA interference and inhibition of MEK-ERK signaling prevent abnormal skeletal phenotypes in a mouse model of craniosynostosis. Nat Genet 2007; 39:1145 - 50; http://dx.doi.org/10.1038/ng2096; PMID: 17694057
- Lomri A, Lemonnier J, Delannoy P, Marie PJ. Increased expression of protein kinase Calpha, interleukin-1alpha, and RhoA guanosine 5′-triphosphatase in osteoblasts expressing the Ser252Trp fibroblast growth factor 2 receptor Apert mutation: identification by analysis of complementary DNA microarray. J Bone Miner Res 2001; 16:705 - 12; http://dx.doi.org/10.1359/jbmr.2001.16.4.705; PMID: 11315998
- Kim HJ, Lee MH, Park HS, Park MH, Lee SW, Kim SY, et al. Erk pathway and activator protein 1 play crucial roles in FGF2-stimulated premature cranial suture closure. Dev Dyn 2003; 227:335 - 46; http://dx.doi.org/10.1002/dvdy.10319; PMID: 12815619
- Opperman LA, Adab K, Gakunga PT. Transforming growth factor-beta 2 and TGF-beta 3 regulate fetal rat cranial suture morphogenesis by regulating rates of cell proliferation and apoptosis. Dev Dyn 2000; 219:237 - 47; http://dx.doi.org/10.1002/1097-0177(2000)9999:9999<::AID-DVDY1044>3.0.CO;2-F; PMID: 11002343
- Opperman LA, Chhabra A, Cho RW, Ogle RC. Cranial suture obliteration is induced by removal of transforming growth factor (TGF)-beta 3 activity and prevented by removal of TGF-beta 2 activity from fetal rat calvaria in vitro. J Craniofac Genet Dev Biol 1999; 19:164 - 73; PMID: 10589398
- Ko SH, Behr B, Longaker MT. Discussion. TGF-beta1 RNA interference in mouse primary dura cell culture: downstream effects on TGF receptors, FGF-2, and FGF-R1 mRNA levels. Plast Reconstr Surg 2009; 124:1474 - 6; http://dx.doi.org/10.1097/PRS.0b013e3181b989de; PMID: 20009833
- Gosain AK, Machol JA 4th, Gliniak C, Halligan NL. TGF-beta1 RNA interference in mouse primary dura cell culture: downstream effects on TGF receptors, FGF-2, and FGF-R1 mRNA levels. Plast Reconstr Surg 2009; 124:1466 - 73; http://dx.doi.org/10.1097/PRS.0b013e3181b98947; PMID: 20009832
- Whitehead KA, Langer R, Anderson DG. Knocking down barriers: advances in siRNA delivery. Nat Rev Drug Discov 2009; 8:129 - 38; http://dx.doi.org/10.1038/nrd2742; PMID: 19180106
- Cooper GM, Usas A, Olshanski A, Mooney MP, Losee JE, Huard J. Ex vivo Noggin gene therapy inhibits bone formation in a mouse model of postoperative resynostosis. Plast Reconstr Surg 2009; 123:Suppl 94S - 103S; http://dx.doi.org/10.1097/PRS.0b013e318191c05b; PMID: 19182668
- Cooper GM, Curry C, Barbano TE, Burrows AM, Vecchione L, Caccamese JF, et al. Noggin inhibits postoperative resynostosis in craniosynostotic rabbits. J Bone Miner Res 2007; 22:1046 - 54; http://dx.doi.org/10.1359/jbmr.070410; PMID: 17437358
- Lajeunie E, Heuertz S, El Ghouzzi V, Martinovic J, Renier D, Le Merrer M, et al. Mutation screening in patients with syndromic craniosynostoses indicates that a limited number of recurrent FGFR2 mutations accounts for severe forms of Pfeiffer syndrome. Eur J Hum Genet 2006; 14:289 - 98; http://dx.doi.org/10.1038/sj.ejhg.5201558; PMID: 16418739
- Rossi M, Jones RL, Norbury G, Bloch-Zupan A, Winter RM. The appearance of the feet in Pfeiffer syndrome caused by FGFR1 P252R mutation. Clin Dysmorphol 2003; 12:269 - 74; http://dx.doi.org/10.1097/00019605-200310000-00012; PMID: 14564217
- Muenke M, Schell U, Hehr A, Robin NH, Losken HW, Schinzel A, et al. A common mutation in the fibroblast growth factor receptor 1 gene in Pfeiffer syndrome. Nat Genet 1994; 8:269 - 74; http://dx.doi.org/10.1038/ng1194-269; PMID: 7874169
- Reardon W, Winter RM, Rutland P, Pulleyn LJ, Jones BM, Malcolm S. Mutations in the fibroblast growth factor receptor 2 gene cause Crouzon syndrome. Nat Genet 1994; 8:98 - 103; http://dx.doi.org/10.1038/ng0994-98; PMID: 7987400
- Slaney SF, Oldridge M, Hurst JA, Moriss-Kay GM, Hall CM, Poole MD, et al. Differential effects of FGFR2 mutations on syndactyly and cleft palate in Apert syndrome. Am J Hum Genet 1996; 58:923 - 32; PMID: 8651276
- Bochukova EG, Roscioli T, Hedges DJ, Taylor IB, Johnson D, David DJ, et al. Rare mutations of FGFR2 causing apert syndrome: identification of the first partial gene deletion, and an Alu element insertion from a new subfamily. Hum Mutat 2009; 30:204 - 11; http://dx.doi.org/10.1002/humu.20825; PMID: 18726952
- Ibrahimi OA, Chiu ES, McCarthy JG, Mohammadi M. Understanding the molecular basis of Apert syndrome. Plast Reconstr Surg 2005; 115:264 - 70; PMID: 15622262
- Cornejo-Roldan LR, Roessler E, Muenke M. Analysis of the mutational spectrum of the FGFR2 gene in Pfeiffer syndrome. Hum Genet 1999; 104:425 - 31; http://dx.doi.org/10.1007/s004390050979; PMID: 10394936
- Rutland P, Pulleyn LJ, Reardon W, Baraitser M, Hayward R, Jones B, et al. Identical mutations in the FGFR2 gene cause both Pfeiffer and Crouzon syndrome phenotypes. Nat Genet 1995; 9:173 - 6; http://dx.doi.org/10.1038/ng0295-173; PMID: 7719345
- Slavotinek A, Crawford H, Golabi M, Tao C, Perry H, Oberoi S, et al. Novel FGFR2 deletion in a patient with Beare-Stevenson-like syndrome. Am J Med Genet A 2009; 149A:1814 - 7; http://dx.doi.org/10.1002/ajmg.a.32947; PMID: 19610084
- Meyers GA, Orlow SJ, Munro IR, Przylepa KA, Jabs EW. Fibroblast growth factor receptor 3 (FGFR3) transmembrane mutation in Crouzon syndrome with acanthosis nigricans. Nat Genet 1995; 11:462 - 4; http://dx.doi.org/10.1038/ng1295-462; PMID: 7493034
- Agochukwu NB, Solomon BD, Gropman AL, Muenke M. Epilepsy in Muenke syndrome: FGFR3-related craniosynostosis. Pediatr Neurol 2012; 47:355 - 61; http://dx.doi.org/10.1016/j.pediatrneurol.2012.07.004; PMID: 23044018
- Doherty ES, Lacbawan F, Hadley DW, Brewer C, Zalewski C, Kim HJ, et al. Muenke syndrome (FGFR3-related craniosynostosis): expansion of the phenotype and review of the literature. Am J Med Genet A 2007; 143A:3204 - 15; http://dx.doi.org/10.1002/ajmg.a.32078; PMID: 18000976
- Jin M, Yu Y, Qi H, Xie Y, Su N, Wang X, et al. A novel FGFR3-binding peptide inhibits FGFR3 signaling and reverses the lethal phenotype of mice mimicking human thanatophoric dysplasia. Hum Mol Genet 2012; 21:5443 - 55; http://dx.doi.org/10.1093/hmg/dds390; PMID: 23014564
- Wallis D, Lacbawan F, Jain M, Der Kaloustian VM, Steiner CE, Moeschler JB, et al. Additional EFNB1 mutations in craniofrontonasal syndrome. Am J Med Genet A 2008; 146A:2008 - 12; http://dx.doi.org/10.1002/ajmg.a.32388; PMID: 18627045
- Bonaventure J, El Ghouzzi V. Molecular and cellular bases of syndromic craniosynostoses. Expert Rev Mol Med 2003; 5:1 - 17; http://dx.doi.org/10.1017/S1462399403005751; PMID: 14987407
- Foo R, Guo Y, McDonald-McGinn DM, Zackai EH, Whitaker LA, Bartlett SP. The natural history of patients treated for TWIST1-confirmed Saethre-Chotzen syndrome. Plast Reconstr Surg 2009; 124:2085 - 95; http://dx.doi.org/10.1097/PRS.0b013e3181bf83ce; PMID: 19952666