Abstract
Signaling pathways that are activated upon interaction of glial cell-line derived neurotrophic factor (Gdnf), its coreceptor Gfrα1, and receptor tyrosine kinase Ret are critical for kidney development and ureter maturation. Outside the kidney, this pathway is implicated in a number of congenital diseases including Hirschsprung disease (intestinal aganglionosis, HSCR) and hereditary cancer syndromes (MEN 2). Total lack of Gdnf, Gfrα1 or Ret in mice results in perinatal lethality due to bilateral renal agenesis or aplasia. In humans, RET mutations have been identified in a spectrum of congenital malformations involving the RET axis including isolated HSCR, isolated congenital anomalies of kidney or urinary tract (CAKUT), or CAKUT and HSCR together. The molecular basis for these pleiotropic effects of RET has just begun to be unraveled. In an effort to delineate the pathogenetic mechanisms that underlie these congenital malformations, we and others have characterized Ret’s role in early kidney and urinary system development. Here we present a brief overview of the “many faces” of Ret dysfunction in kidney with particular emphasis on Ret’s signaling specificity and intergenic interactions that confer normal urinary system development.
Clinical Aspects of Congenital Urinary Tract Anomalies
Congenital anomalies of kidney or lower urinary tract (CAKUT) are common anomalies (1 in 250 live births) and the most common cause of renal failure in children.Citation1 These include renal hypoplasia/dysplasia, agenesis, hydronephrosis, mega-ureter/hydroureter, duplicated collecting system, abnormal valves and distal obstruction. The abnormalities can occur in isolation, or as a complex, or as a part of other syndromes involving multiple organ systems. In some cases they are incompatible with life and even after surgical correction, many patients progress to renal failure or have associated comorbidities that include skeletal, cardiovascular and hematopoeitic systems. Thus, there is considerable interest in understanding molecular genetics of CAKUT and related diseases. In many respects rodent kidney development is similar to human kidney development. For example, kidney development in both occurs in an anterior to posterior direction in three stages, pronephros, mesonephros and metanephros (see below). Further, several genes have been identified that regulate kidney development in both humans and rodents. Among these alterations, mutations in RET, a receptor tyrosine kinase (RTK), were recently found in about 35% of patients with various forms of renal agenesis.Citation2 In an effort to better understand how RET might lead to CAKUT, several laboratories have characterized Ret mutant animal models and deciphered the various processes during mammalian kidney development that depend on Ret, and in doing so have provided valuable insights into kidney organogenesis and CAKUT pathogenesis. Today I will provide a brief overview of the diverse roles of Ret pathway in early kidney development with particular emphasis on insights gleaned about the basis for signaling specificity from animal models.
Urinary System Development
There are three pairs of embryonic kidneys that develop in mammals in an anterior to posterior direction, the pronephros, mesonephros and metanephros.Citation3 First, the pronephros develops from a collection of cells in the intermediate mesoderm (IM) that undergo mesenchymal to epithelial transition and initiate the development of nephric duct (Wolffian duct, WD) (in mice at about E8.5d; in humans at 3 weeks of gestational age). The pronephros is a transient, non functional structure that is followed by another transient structure, the mesonephros (at about E9d in mice; about 25d gestation in humans). The mesonephros contains a linear organization of nephrons along the growing WD. In mice only the rostral/anterior mesonephric tubules are connected to the WD. These later become part of the epididymis in males. The WD becomes the vas deferens.
Development of the definitive kidney or metanephros in mice begins at E10.5–E11.5 as an outgrowth from the WD known as ureteric bud (UB, future collecting system).Citation4,Citation5 Specific signals from the metanephric mesenchyme (MM) regulate UB outgrowth into MM, a process called induction. Upon reaching the MM, the UB tip undergoes branching morphogenesis to form the collecting system. The initial UB stalk becomes the ureter. The branching UB tips reciprocally induce the surrounding mesenchyme (causing mesenchymal condensation), a process that stimulates nephrogenesis. At the same time, equally important events occur in the distal WD and the ureter that determine proper connection of the ureter to the bladder. The ureter undergoes remodeling which involves separation from the WD (future genital duct in males), insertion and migration into the trigone accompanied by common nephric duct (CND) degeneration.Citation6 Abnormal ureteral maturation results in megaureter, hydronephrosis, vesico-ureteral reflux, uretero-pelvic junction (UPJ) abnormalities and obstruction, while abnormal UB induction results in renal agenesis, hypoplasia or dysplasia. More severe abnormalities in these processes will present in utero or early childhood and many result in renal failure in children. Less severe abnormalities such as mild hypoplasia (suggesting reduced branching morphogenesis and nephrogenesis) may remain undetected by conventional screening procedures, and may result in hypertension and chronic renal failure.
Diseases Associated with RET
RET is an acronym for “rearranged during transfection.” It was discovered as a gene-rearrangement that resulted in transformation of NIH3T3 fibroblasts when human lymphoma DNA was transfected into these cells.Citation7 RET encodes a single pass, transmembrane receptor tyrosine kinase (RTK) that resides at chromosome 10q11.2 in the human genome.8 It consists of 20 exons with a potential to generate two major isoforms due to alternative splicing in the carboxy terminus () (see below).Citation9 RET homologues exist in all vertebrates and have also been identified in Drosophila melanogaster. RET mutations have been identified in several human diseases. Activating RET mutations predispose to hereditary oncogenic syndromes such as multiple endocrine neoplasia type 2 (MEN2), are present in 30–70% of sporadic medullary thyroid carcinomas, and have been identified in papillary thyroid carcinomas. More recently RET and estrogen receptor pathway interaction was shown to be important in development of breast cancer.Citation10 Inactivating RET mutations are a predominant feature of hereditary Hirschsprung disease (HSCR, intestinal aganglionosis) and also found in a large number of sporadic HSCR.Citation11 Majority of gain and loss of RET phenotypes known to date present in an autosomal dominant manner. RET mutations have been reported in patients presenting with MEN2 and HSCR, CAKUT and MEN2, and more recently in patients with isolated CAKUT, or with HSCR and CAKUT.Citation2,Citation12,Citation13 These observations illustrate the potential for aberrations in RET signaling to predispose for both isolated as well as multisystem defects in urinary and other systems. They have led to increased efforts to understand the mechanisms of RET function at cellular as well as at organ level in hopes of delineating how abnormalities of RET signaling can be associated with multiple syndromes.
Gross Anatomy of RET
RET consists of three main domains, extracellular, transmembrane and cytoplasmic.Citation14 The extracellular domain plays a role in receptor dimerization and interaction with one of its coreceptors and ligand (). While the predicted molecular mass of RET is 150 kDa, glycosylation in the amino-terminus results in an approximately 170 kDa mature protein. The tyrosine kinase domain resides in the cytoplasmic portion. There are two major isoforms of RET, RET9 (1072aa) and RET51 (1114aa). These are generated by alternative splicing in the 3-prime (3′) region of RET such that the carboxy terminus of the mature protein is identical until residue 1063. Thus RET9 has nine amino acids in the carboxy terminus that are different than the 51 terminal residues in RET51.
GDNF-GFRα1-RET Signaling
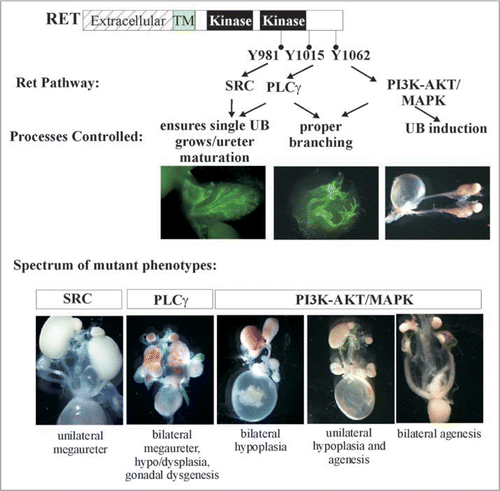
Glial cell line-derived neurotrophic factor (GDNF) family ligands (GFLs) are a family of four neurotrophic factors, GDNF, Neurturin, Artemin and Persephin (). Interaction of a homodimer of one of the GFLs with a homodimer of one of their preferred coreceptors (GFRα1-4), increases affinity of GDNF-GFRα1 to interact with a RET homodimer.Citation15 This results in a multicomponent activated receptor complex leading to phosphorylation of key tyrosine (Y) residues in the cytoplasmic domain. RET-Y905 is a site necessary for kinase activity and modulates autophosphorylation of four key docking tyrosines that serve as binding sites for intracellular adaptors leading to activation of major signaling pathways. Activation of these pathways determines the type of biological response to be elicited in a cell. RET-Y905 regulates binding to Grb7/10, RET-Y981 binds to SRC, RET-Y1015 to PLCγ, and RET-Y1096 which is only present in RET51, to GRB2. Further, RET-Y1062 serves as a multi-docking adaptor site for several intracellular adaptors including SHC, FRS2, IRS1/2, DOK1, DOK4-6 and ENIGMA (). Phosphorylation at RET-Y1062 enables binding to adaptors like SHC to activate PI3K/AKT or MAPK cascades through recruitment of GRB2/GAB or GRB2/SOS, respectively. The PI3K/AKT pathway can also be activated by direct recruitment of GRB2-GAB complex on phospho-RET-Y1096 present in RET51. The major pathways activated by SRC and PLCγ are RAS-MAPK and PKC, respectively. These pathways regulate cellular processes such as proliferation, migration, survival and regeneration in a number of tissue specific stem cells/progenitors including the spermatogonial stem cells, enteric neural crest precursors, CNS and PNS neurons, and in the embryonic kidney (metanephros).Citation16 One active area of investigations is deciphering the biological need for the various RET isoforms and the signaling cascades in kidney development and disease. Do they provide redundancy or specificity?
Gdnf-Gfrα1-Ret Expression in the Kidney and Lower Urinary Tract
Much of our understanding of the role of Ret signaling pathway in kidney development and function comes from the knowledge of spatio-temporal expression patterns of Ret signaling components in conjunction with phenotypic analysis of various Gdnf/Gfrα1/Ret mutant mice. Using in situ hybridization or analysis of reporter mice that express lacZ or green fluorescent protein (GFP) from Ret or Gfrα1 loci we have detected Ret and Gfrα1 expression in the developing WD in the pronephric kidney as early as E8.5d in mice () (data not shown). Before UB budding at E10.5, increased Ret expression is detected in the distal WDs suggesting its role in UB induction.Citation17–Citation19 In mice, expression of Gdnf from the MM at about E10.5 activates Gfrα1-Ret signaling in the WD to promote UB outgrowth into the MM and subsequent branching morphogenesis.Citation20 During branching morphogenesis, Gdnf expression becomes more localized to the undifferentiated mesenchyme in the periphery and it is markedly reduced or undetectable in the mature kidney.Citation4 The postnatal expression of GFLs is poorly defined in the kidney. Among the other GFLs, Neurturin is also expressed in the UBs of embryonic kidney after E14. However, its physiological relevance is not known as Neurturin null animals have normal kidneys.Citation17,Citation21 In the initial stages of metanephric kidney development both Ret and Gfrα1 are expressed in the ureteric bud stalk and the UB tip. During branching morphogenesis Ret and Gfrα1 expression diminishes in the UB stalk and their expression is more confined to proliferating UB tip cells. Towards the later stages of kidney development, high expression of both these genes is seen in the nephrogenic zone. Postnatal expression of Ret and Gfrα1 is less defined, although our recent observations using Gfrα1 and Ret reporter mice detect Ret and Gfrα1 in collecting ducts, with weaker expression in older mice (unpublished observations). Interestingly, Gfrα1 expression in the kidney is broader than Ret as it is also detected in the cap mesenchyme surrounding the branching UB tips and in a subset of cortico-medullary proximal tubules in adult kidneys (unpublished observations).Citation20,Citation22,Citation23 Gfrα1 expression in structures that don't express Ret has been attributed to Ret-independent roles or in trans-Ret activation.Citation24 Although, trans expression is dispensable for normal kidney development, it may have unrecognized roles in adult kidney under pathological conditions.Citation23
Biological Roles of Gdnf-Gfrα1-Ret in Kidney Development
Consistent with these expression patterns of Gdnf, Gfrα1 and Ret in the developing kidney, gene knock-out studies reveal that this pathway is essential for early kidney development as the absence of Gdnf, Gfrα1 or Ret during embryogenesis in mice results in renal agenesis/rudimentary kidneys and perinatal lethality.Citation18,Citation25–Citation29 Several findings suggest that Ret signaling also has a role in later kidney branching morphogenesis. These include expression of Ret signaling components in later kidney development, inhibition of branching in vitro using Ret-activated pathway inhibitors, and dependence of branching on stimulating or inhibiting Gdnf.Citation30–Citation32 However, direct proof for Ret's role post-UB induction in vivo would require its conditional inactivation after early UB induction has occurred.
The agenesis phenotype in Gdnf/Gfrα1/Ret null animals suggested a role in early UB induction and growth. A series of innovative studies to illuminate Ret's role at a cellular level in orchestrating branching morphogenesis were undertaken by Dr. Costantini's laboratory. Chimeric animals were generated in which “trackable” Retnull cells and wild-type cells contributed to the kidney. It was observed that during branching these Ret-null cells never populated the UB tip, showing that intact Ret signaling is required for populating the UB tip.Citation33 More recently, in a similar series of experiments it was shown that Ret activity was necessary to modulate WD epithelial cell rearrangements at the distal WD prior to UB budding.Citation34
To determine if Gdnf in MM provides tropic cues to the UB to reach the MM and branch, Shakya et al. expressed Gdnf exclusively in the WD by expressing Gdnf under the regulation of HoxB7 promoter in the background of Gdnf null mice.Citation35 They observed that generalized expression of Gdnf only in WD and the urinary tree was able to support metanephric kidney development. However, ectopic UBs also develop, indicating that expression of Gdnf in MM is not necessary for the UB to reach the MM and undergo branching morphogenesis. This suggests that there are redundant cues in MM for UB tropism.
Gdnf, Gfrα1, Ret Signaling in the Lower Urinary Tract
Formation of the ureterovesicular junction is a critical event in ensuring the proper union and communication between the upper (derived from WD) and lower urinary tracts (cloaca). It requires that distal portion of WD inserts normally in the cloaca (by E10.5), followed by CND degeneration (by E14.5), ureter separation from the WD and its migration into the trigone to connect to the bladder lumen.Citation6,Citation36,Citation37 Defects in this process can result in failure of the ureter to connect or traverse through the bladder and induce reflux nephropathy. In children this is one of the most common causes of renal failure. Degeneration of the CND is a critical event for proper connection of the upper and lower urinary tracts and development of the male genital tract. Ret expression in the CND is markedly increased at E11.5 and both Ret and Gfrα1 are expressed in the CND until CND degenerates by E14.5. Gdnf is expressed in the surrounding urinary bladder wall. Consistent with this expression, a number of ureter abnormalities are present in Ret-null mice and include unilateral/bilateral ureter agenesis, blind ending ureters and failure to connect to the urogenital sinus.Citation6,Citation18 These studies suggest that Ret is likely involved in promoting CND apoptosis and ensuring proper connection of the upper and lower urinary tracts. While upstream pathways such as retinoic acid receptor signaling positively regulate Ret in ureter maturationCitation6,Citation36 it was reported recently that members of the LAR family of protein tyrosine phosphatases negatively regulate Ret signaling in the formation of UVJ by controlling distal ureter apoptosis.Citation38 Because UVJ abnormalities are a major cause of CAKUT, it is important to delineate specific pathways emanating from Ret or the CND niche that regulate ureter remodeling.
Insights into CAKUT from Animal Models with Altered Ret Signaling
While studies performed using Gdnf, Gfrα1, Ret null mice revealed an absolute requirement for Ret signaling in kidney development it wasn't until Ret-mutants with severely diminished signaling were characterized that a wider role of Ret in kidney development was defined. One of these mutants expressed a Ret hypomorphic allele that in homozygous state causes intestinal aganglionosis and hypoplastic, cystic kidneys.Citation39 Another example is a mouse that expresses a Ret dominant negative allele, that severely diminishes signaling through Ret-Y1062 (Shc binding site and activate PI3K/MAPK pathway).Citation40 This mouse presents with varying degrees of abnormal kidneys including bilateral hypoplasia, unilateral hypoplasia and unilateral agenesis (). Many of these mice survive for several weeks postnatally and a subset of the mice develops renal cysts. Eventually these mice fail to thrive and die due to varying degrees of small and large bowel aganglionosis. Results from these studies not only created useful animal disease models, but also provided a glimpse of wider roles of Ret pathways in kidney development especially insights into how varying activities of Ret are associated with a spectrum of renal malformations, Ret's potential role in postnatal collecting duct cystic abnormalities, and how differential Ret activity can impact development of the urinary and other systems.
While my lecture focused on Ret and downstream signaling in kidney development, it should be noted that several factors such as Eyα1, Sal1, Pax2, Six2, Wnt11 or Gdf11 can also modulate Ret signaling and influence kidney branching by altering Gdnf expression in the mesenchyme or by modulating the availability of Gdnf to Ret such as by 2-O sulfation.Citation5,Citation41 Animals with mutant forms of these genes present with a range of kidney defects due to aberrant Ret signaling.
The Role of Specific Ret-Activated Pathways in CAKUT Pathogenesis
Ret activates several major signaling cascades and exists in at least two major isoforms. To ascertain whether each makes an incremental contribution towards UB induction, or whether each has a specific role, we and others have undertaken studies to examine the impact of individual Ret pathway mutants and isoforms on organogenesis. One of the questions examined was the biological relevance of the extra Grb2 docking site that preferentially activates PI3K/AKT pathway in the Ret51 isoform. Two groups pursued this question by characterizing mice that express exclusively either the Ret9 or Ret51 isoform. Interestingly, quite different results were obtained. A mouse expressing only Ret51 where Ret51 was a chimeric form consisting of mouse extracellular-human cytoplasmic domain was not able to fully support normal kidney or ENS development.Citation39 This suggests that the carboxy-terminus of Ret51 perhaps has an inhibitory role that could be either due to the different nine residues in the two isoforms or due to the additional Grb2 binding site. In contrast, when mice were made that exclusively express human wild-type RET9 or RET51 isoforms, it was found that either isoform is competent for proper kidney development.Citation42 Further, mice in which the nine residues after RET1063 in RET9 and RET51 were swapped also did not manifest differences in kidney development. This suggests that the extra or different residues in carboxy-terminus between RET9 and RET51 are of no significance for normal development and that the portion of RET51 containing the Grb2 binding site is dispensable.Citation42 The reasons for the different results between the two groups are not entirely clear. However, the findings raise the possibility that in different contexts the isoforms could have redundant or unique roles.
To examine the biological roles of each of Ret's major tyrosine adaptor docking sites mice were made that transduce signals from all docking sites except one.Citation42 This was accomplished by the generation of mice that only express mutations (Tyr-Phe) in Src, Plcγ or Shc adaptor sites or that lack the carboxy terminal 43 residues in RET51 with the Grb2 site. The results show that only the Plcγ mutants (RET51-Y1015F) were not able to support proper kidney development (). In fact, abrogation of the Plcγ site resulted in a quite complex CAKUT phenotype with bilateral megaureters, hypoplastic and dysplastic multicystic kidneys, failure of separation of gonadal duct from the ureter leading to attachment of gonads to the urinary tree in both males and females, and possibly distal ureter abnormalities (). Characterization of these mutants during embryogenesis revealed that the phenotype is partly related to presence of supernumerary ureters and partly due to branching defects. However, additional studies during mesonephric and early metanephric kidney development will likely provide insights into the developmental basis for these results including defining whether UB sprouts from the right position, and problems associated with mesonephros degeneration, insertion of WD into cloaca and CND degeneration. This new phenotype associated with abrogation of only one specific signaling cascade defines yet another role of Ret in CAKUT that could not have been realized by knock-out studies.
To further explore whether redundant signaling from Grb2 masks essential roles of the Src or the Shc adaptor site, mice with mutations in Plcγ, Src and Shc binding sites were made in context of the RET9 isoform.Citation42 The results in general show that the Grb2 containing carboxy terminus provides redundancy to both the Src (RET9-Y981F) and multidocking Shc (RET9-Y1062F) sites, although the impact at these sites appear to be different. For instance, the RET9-Y981F mutants show incomplete penetrance of CAKUT with only a subset harbor megaureters. On the other hand loss of Shc binding in the face of absent Grb2 resulted in phenotype quite similar to Ret-null mice, bilateral agenesis or kidney rudiments (). The same result was also seen in mice expressing a chimeric Ret9-Y1062F allele made by fusion of mouse Ret extracellular and human Ret intracellular domains.Citation43 A different approach to examine the role of the Shc binding site was taken by creating this mutation in the endogenous mouse Ret (i.e., it would express this mutation in both Ret9 and Ret51). This resulted in slightly hypoplastic kidneys.Citation44 Together these studies suggest that Grb2-mediated signals provide redundancy to Src and Shc binding sites and totally abrogating Grb2 (through Shc or direct Grb2 binding) is the reason for agenesis in Ret-null mice. Interestingly, the role of Grb2 on the Src site in kidney development seems to be different than on the Shc site as Ret-Src mutants show a megaureter phenotype as opposed to agenesis typically seen in all Ret9 mutants deficient in Shc binding. These animal models provide remarkable insights into specificity of Retactivated signaling cascades that determine the manifestation of different types of CAKUT.Citation40,Citation42–Citation44
Genetic Interactions of Ret Signaling Complexes that Influence Kidney Development
Both positive and negative regulators influence kidney development. These include activating factors upstream, either in the MM or in mesonephric mesoderm influencing Ret activity through Gdnf or in the UB, and downstream of Ret such as Wnt11 (forms a positive feedback loop), and repressors such as Spry in the WD or metanephros.Citation45 I will briefly review examples where direct evidence of genetic interactions between Ret signaling complex and other genes has been observed in vivo. One of the first examples of this came from studies of Spry1 null and Gdnf heterozygous animals. Without Spry1, mice develop a CAKUT phenotype with features of bilateral megaureters, gonadal abnormalities and multiplexed hypoplastic/dysplastic kidneys.Citation46 Because these mice have supernumerary ureters and Gdnf in kidney organ cultures can induce extraneous UB budding and ectopic ureters, it was suggested that perhaps Gdnf was aberrantly expressed leading to the observed phenotype. Further support for this concept comes from several mouse mutants that have supernumerary ureters due to failure to limit Gdnf expression to posterior IM.Citation47,Citation48 However, Basson et al. did not detect abnormal anterior Gdnf expression in the IM in Spry1 null mice suggesting that WD in the Spry1 null mice is more responsive to Gdnf. They supported this idea by breeding Spry1 homozygous mice onto Gdnf heterozygous background and found that in a majority of double homozygous/heterozygous, the CAKUT phenotype appears to normalize, meaning Gdnf haploinsufficiency almost cures CAKUT in Spry1 null mice.Citation46 Basson et al. also demonstrated that Spry1 signaling was important in kidney branching, and this was dependent on Spry1 interactions with Gdnf-Ret signaling.Citation49 However, they showed that the mechanism of Spry1 interaction with the Ret pathway during branching was different than its role in the WD. In UB branching Gdnf expression was much wider in MM in Spry1 nulls, whereas in WD no increased or wider Gdnf expression in the anterior IM was noted. The increased Gdnf in MM was likely due to increased Wnt11 expression in UB as it induces Gdnf expression in MM. Reducing Gdnf dosage also normalized the branching defects in Spry1 null animals.
Spry proteins inhibit the MAPK cascade.Citation50 MAPK activation by Ret is primarily dependent on signals emanating from Ret-Y1062, a residue important for UB induction. We tested whether the Spry1 CAKUT phenotype will normalize in the background of RET9-Y1062F mutants.Citation51 We observed that double homozygous Spry1:RET9-Y1062F mutant mice have essentially normal kidneys providing in vivo evidence that Ret and Spry1 genetically interact ().
This result has several important implications. First, the ability to normalize two remarkably different defects, renal agenesis in RET9-Y1062F and the complex CAKUT phenotype in Spry1 null, by expressing them in the same genetic background may be applicable in correcting these disparate defects in fetuses by modulating these individual signaling pathways. Second, it provides evidence for the importance of negative regulators in controlling RTK-mediated kidney development. Third, it provides support to the possibility that RTKs other than Ret that may play a role in kidney development but are normally repressed by Spry1 (see below). Studies from Basson et al. and Rozen et al. provide a model where without Spry1, there is RTK overdrive that leads to extra UB outgrowths especially in the mesonephric duct. This is mainly dependent on signals from Ret-Y1062 which in the presence of wild-type levels of Spry1 are somewhat dampened in the anterior WD and therefore do not produce supernumerary ureters. Without Spry1, Ret-Y1062 dependent activation of MAPK is at a much higher level producing ectopic ureters since abrogation of Ret-Y1062 signaling in Spry1 null does not produce ectopic ureters. The fact that UB normally grows from distal WD in double Ret9-Y1062F: Spry1 null animals suggests that other RTKs or other Ret-dependent signaling pathways must be compensating in these double mutants to produce normal kidneys since the main inducer of UB, Ret-Y1062 signaling, has been abrogated (). One of these pathways appears to be Fgf signaling. The role of signaling by Fgf receptors as an alternate pathway for early UB outgrowth was suggested by studies where aberrant expression of soluble Fgfr2IIIb resulted in renal agenesis.Citation52 Chi et al. demonstrated interactions between Gdnf, Fgf7, Wnt11 and Spry signaling by showing that renal agenesis in transgenic mice expressing human SPRY2 in the UB could be rescued by Gdnf or Fgf7, supporting that Fgf signaling has the potential for stimulating UB and branching.Citation45 Recently, Maeshima et al. showed that Fgf7, induced UB outgrowth in Ret null WDs when activin signaling was inhibited.Citation53 Conclusive evidence for participation of other RTKs in the network of Ret-Spry1 in the early events of WD growth to sculpt the metanephric kidney would require generation of multiple mutant alleles including Fgf pathway mutants, Spry1 and Ret signaling mutants.
An important observation from the Spry1:RET9-Y1062F mice is that the kidney defects were normalized but the intestinal aganglionosis was not (). So in essence these double mutants manifest as HSCR only even though they harbor detrimental mutations in RET known to severely affect kidney development when present in isolation. Thus, depending on the combination of the RET and SPRY1 mutations inherited/expressed, one could be predisposed to CAKUT alone (SPRY1 deficiency), renal agenesis and HSCR (RET9-Y1062F mutant), or HSCR alone (double Spry1;Ret homozygous mutant mice). These organ specific genetic interactions may explain some cases of HSCR where RET mutations in the cytoplasmic domain may not have any overt renal anomalies or the basis for coexistence of CAKUT and HSCR. It would be important to delineate other putative modifiers of Ret mutant phenotypes to get a better understanding of organ specificity in disease manifestation. Further, because of possible genetic interactions, patients with any of the diseases related to RET, such as HSCR, MEN2 or CAKUT, presenting alone or in combination, may benefit by screening for mutations in RET and other genes that modify RET's biological responses.
Genetics of CAKUT
Recent advances in genetics and analysis of a number of gene-deficient mouse models that lead to renal malformation have led investigators to examine the implications of their findings in humans with CAKUT. These studies point to strong associations between genes deemed important from animal studies and the identification of mutations in these in CAKUT. For example, Robo2 disruption results in megaureters with distal obstruction and dysplastic kidneys.Citation47 Sequencing of ROBO2 in CAKUT samples identified a subset of VUR patients with mutations that disrupt ROBO2 function.Citation54 Other genes that have roles in urinary system development and have been found to be associated with mutations in patients with isolated kidney defects or in syndromes include BMP4, FOXC1, ACE, Chr13q33–34, GATA3, PAX2, EYA1, SAL1 and SIX1.Citation55–Citation58 Animal studies have indicated that many of these influence Gdnf-Ret signaling. Recently, RET coding region mutations were identified in 12 of 33 patients with unilateral or bilateral agenesis.Citation2 Several of these were in cytoplasmic domain of RET suggesting that decreased RET activity in humans results in renal agenesis, as in the numerous Retmutant animals discussed above. Since animal studies now suggest a much larger role of RET in CAKUT, further investigations in larger human cohorts and in a broader spectrum of CAKUT would be needed to determine the incidence of RET mutations in these malformations, and more importantly where or what function of RET is affected in these mutations. Since RET has pleiotropic roles in human diseases including HSCR, MEN2 syndromes, patients presenting with RET mutations in these conditions should be closely monitored for renal defects. CAKUT patients should be monitored for HSCR phenotype as RET intergenic interactions with its positive and negative regulators can influence organotypic specificity of disease manifestation ().
Conclusion
Since the initial discovery that Ret is essential in kidney genesis almost fifteen years ago, efforts of several laboratories have markedly enhanced our understanding of the roles of Ret signaling complex in urinary system development. These efforts have defined new roles of Ret in kidney development through analyses of hypomorphic or defined Ret-signaling mutants and helped characterize how variations in amounts or type of Ret signaling can result in a wide array of CAKUT phenotypes. Besides delineating specificity of Ret-dependent cascades and other positive and negative regulators of Ret in kidney development, the efforts have led to a better understanding of the basic processes of branching morphogenesis, reciprocal interactions, cell proliferation, migration, differentiation and survival, and above all, of kidney and lower urinary tract development. New roles of Ret in distal ureter maturation have been discovered, and insights derived that explain how complex phenotypes that encompass distal ureter abnormalities such as mega-ureter/hydroureter and defects such as renal hypoplasia or dysplasia can develop either together as seen in Ret-Y1015F-Plcγ mutant mice or in isolation as seen in Ret hypomorphic mice. Experiments with genes modifying Ret phenotypes have also provided information that depending on the context, upper and lower urinary tract abnormalities can be detected singly, or together. The relevance of findings from animal Ret mutant models was confirmed by the discovery of high incidence of RET mutations in patients with kidney agenesis.
While significant strides have been made in our understanding of Ret biology and kidney development, several aspects remain to be explored. For example, developmental processes that lead to the complex phenotypes in Ret mutants remain to be defined particularly the events during mesonephric kidney development. Also, it must be determined what specific signals regulate ureter maturation. Since many mutants in positive and negative regulators produce the same phenotypes as Ret mutants, studies that examine which of these interact with Ret in vivo need to be conducted in order to determine which pathways in kidney development converge and which interactions are relevant in kidney versus in other organs that are dependent on Ret for development. Specific questions that should be addressed include the following: Is there specificity of Spry proteins in kidney versus gut development? Do other genes such as Slit2, Bmp4, Foxc1 interact with any of the specific Ret cascades? What are the relevant molecules downstream of Ret that modulate kidney development? Are there any positive or negative feedback mechanisms that are important?
Ret continues to be expressed in developing kidney post-UB induction. Its expression pattern changes from being ubiquitous in the urinary tree to more focal. Deciphering the transcriptome of this Ret-positive population in temporal manner in order to delineate the global changes that occur in these progenitors as kidney development proceeds could identify new molecules that may have important roles in kidney development. Further, studies need to address if developmentally important genes such as Ret have roles in regeneration of adult kidney after injury as increasingly it is being found that molecular signals that are important during early development may have roles in regenerating damaged kidneys after injury.
Defining molecular aberrations leading to CAKUT in Ret mutant animals and in humans will uncover biomarkers that can be used for the early diagnosis of renal abnormalities in children. While finding RET mutations in patients with renal agenesis is certainly a big step in this direction, larger scale studies encompassing a wide spectrum of CAKUT phenotypes, in different populations can define clinically relevant mutations in RET and interacting genes. Further, whole genome-wide studies are needed to discover multigenic causes of CAKUT that may not be apparent from screening of individual target genes.
Questions and Answers
When you breed the SPRY1 knock out with your RET knockout, that results in an interesting rescue. Do you have any idea whether or not there is a proliferative pathway that SPRY1 is using as an alternative to the MAPK kinase pathway, for instance the mTOR pathway?
I don't know of studies where Spry1 modulates mTOR pathway. What is known from literature about SPRY1 is that MAPK pathway inhibition is its major target.Citation59 Initially, Spry members were shown to inhibit Fgf signaling in lung, and more recently evidence has been provided for Spry inhibiting Ret and Fgf pathways in the kidney.Citation45–Citation49 That is why we think that Fgf signaling may play a role. Chi et al.Citation45 also alluded to role of Spry proteins in modulating pathways other than MAPK in kidney, however, the molecular nature of these have not been defined. Dr. Sanjay Nigam from UCSD has also shown that if you treat WD derived from Ret knockout animals with FGF7 in presence of inhibitors of activin you can actually induce UB outgrowth further supporting Fgf as an alternate pathway.Citation53
What is the biochemical basis for the RET signaling? It seems that RET activates PI3 kinase and MAPK kinase, but another kinase pathway upstream (SPRY1-raf) also activates MAPK. What do you think is the phosphorylation site for MAPK kinase downstream of RET? Could it be different from the other one and is that why there are two different phenotypes?
The question is about this idea of convergence of many different cascades into these common signaling pathways. It has always been in an interesting biological question why that occurs. From experiments in vitro it seems to be that they activate MAPK in a similar fashion as the intracellular adaptors that activate Ras-Raf-Erk1/2 are similar. The basis for specificity for different RTKs in part comes from their specific ligands or coreceptors or cell-type specific expression patterns.Citation60 Similarly, Spry proteins are themselves regulated at transcriptional and translational level by RTKs, and in turn they can inhibit Ras-MAPK pathways activated by RTK. This can also vary at cell-type leveI. Thus there is potential for intra RTK as well as inter RTK modulation of MAPK cascades.
It has been proposed in a recent paperCitation34 that migration of Wolffian duct epithelial cells near the caudal part of the Wolffian duct is involved in the formation of the ureteric bud. That process depends on Ret activity. You describe in some of your mutants the formation of multiple budding sites and some are very anterior to the normal site. Do you see any abnormal cell movement associated with the formation of the ectopic buds and what do you think is the positional determinant for these buds?
We haven't really looked at migration in our Ret mutants in the kidney in detail. We do see an increased Ret signal at the distal Wolffian duct before the UB evaginates, which we believe is due to increased proliferation. Studies from the laboratory Dr. Constantini, Columbia University, show this may be due to migration of anterior WD cells in this zone and epithelial rearrangements.Citation34 We have not explored this phenomenon in the ectopic UBs seen in our mutants. Regarding the question of the extra budding from the mesonephric portion of the Wolffian duct, we believe in these regions Spry1 inhibition is likely overcome due to increased Ret expression/signaling, thus tipping the balance in favor of Ret activated MAPK pathways. This was shown by Basson et al. who observed increased MAPK activity in WD with ectopic UBs,Citation46 and in their studies in which Spry1 deletion in UB causes accelerated UB branching.Citation49
Abbreviations
| UB | = | ureteric bud |
| WD | = | wolffian duct |
| MM | = | metanephric mesenchyme |
| IM | = | intermediate mesoderm |
| RTK | = | receptor tyrosine kinase |
| CAKUT | = | congenital anomalies of kidneys or urinary tract |
| HSCR | = | hirschsprung disease |
| MEN | = | multiple endocrine neoplasia |
| Gdnf | = | glial cell line-derived neurotrophic factor |
| UVJ | = | ureterovesicular junction |
| UPJ | = | ureteropelvic junction |
| ENS | = | enteric nervous system |
Figures and Tables
Figure 1 Ret gene structure and signaling. (A) Gene structure of RET. RET is located at chr 10q11.2 and consists of 20 exons (rectangular boxes). Depicted are two major RET isoforms, RET51 and RET9, generated by alternative splicing. (B) Diagram showing RET activation by glial cell line-derived neurotrophic factor (GDNF) family ligands (GFLs). A dimer of one of the four GFLs (GDNF; Neurturin, NRTN; Artemin, ARTN; Persephin, PSPN) associates with homodimers of one of the four GPi-linked corereceptor's (GFRα1–4) to form multimeric complex with a RET homodimer. This leads to activation of RET kinase domain in the cytoplasmic side by phosphorylation, and downstream signaling cascades that regulate the indicated biological responses. (c) Gross anatomy of RET. Different RET domains are shown, extracellular, transmembrane (TM) and cytoplasmic domain that harbors the kinase activity regions. Key RET tyrosine (Y) residues, the preferred intracellular adaptors that they bind to, and the downstream signaling cascades that are activated are shown. Y905 is a residue important for RET kinase activity. Note the extra Tyr Y1096 in the RET51 isoform directly binds to GRB2 to preferentially activate PI3K/AKT pathway. Thus, both RET-Y1062 and RET-Y1096 can activate PI3K through GRB2-GAB1/2.
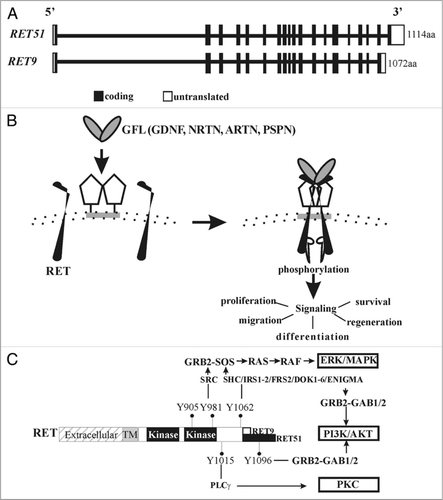
Figure 2 Ret expression in early developing murine kidney. Cartoon shows Ret expression in relation to early stages of embryonic (E) murine kidney development. Wolffian duct (WD) is represented as vertical tube, ureteric bud (UB) as an outgrowth from caudal end of WD, common nephric duct (CND) at distal end of WD, and metanephric mesenchyme (MM) expressing Gdnf as red oval. Ret is first detected at E8.5 in cellular aggregates in the pronephros, then continues to be expressed in the growing WD during mesonephros development, albeit at lower levels. Intense Ret expression is seen at the beginning of metanephric kidney at distal end where UB develops at E10.5. When UB reaches MM (at E11.5), first branching event occurs and intense Ret expression is observed in the tips of the UB. As branching continues, strong Ret expression is confined to UB tips, and Ret expression in WD becomes almost undetectable except in the CND, and becomes weaker in the UB stalk. The intensity of blue correlates with Ret expression.
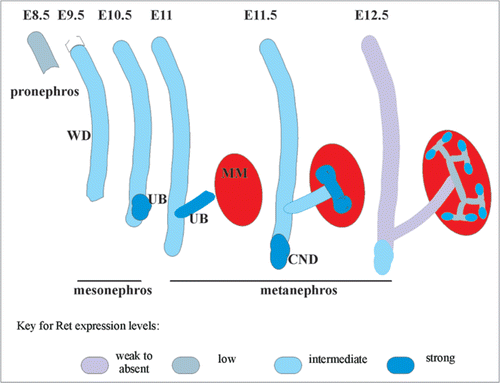
Figure 3 Hypomorphic Ret allele provides evidence that varied Ret signaling is associated with different CAKUT manifestations. (Top row), Compared with wild-type (wt) mice, RetDN/+ mice (express a Ret form with severely reduced, but not absent, Ret-Y1062 signaling) showed a spectrum of renal defects including bilateral small kidneys and unilateral renal agenesis (k, kidney; a, adrenal; ub, urinary bladder, t, testis; o, ovary; adrenal gland, arrowhead). (Bottom row) Bar graph shows decreased total number of glomeruli in RetDN/+ kidney at birth (n = 3 animals, 5 kidneys) compared with wild-type litter mates (n =3 animals, 6 kidneys) (*p < 0.001, mean ± s.e.m). H&E-stained sections show tubulocystic degeneration (arrows) in approximately 50% of 3- to 4-week-old RetDN/+ mice (6 of 12) (co, cortex; me, medulla; pe, pelvis). Scale bar = 5 mm in top row, and 600 µm in bottom row. The figure is adapted from Jain et al.Citation40 with permission.
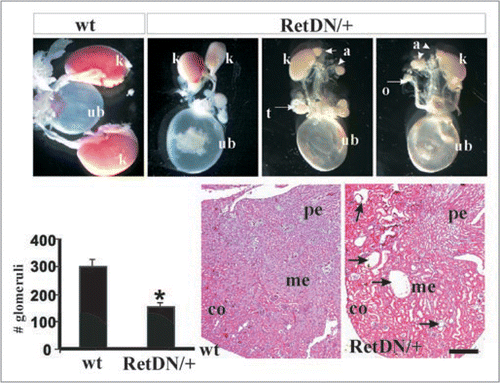
Figure 4 Roles of Ret isoforms and downstream cascades in kidney development. (A) Schematic of the different wild-type RET9, RET51 and their respective mutant knocked-in alleles. The different domains and sizes of the RET9 and RET51 alleles are indicated. The red and blue colors represent the area of divergence between the two RET isoforms. Also indicated are the key docking tyrosine (Y) residues. Homozygous mice were generated that harbor Tyr→Phe (Y→F) for each of the indicated Tyr except Y1096. RET51(Cdel) is an allele where residues 1063–1072 of RET9 were replaced with residues 1063–1072 of RET51 (results in receptor that is essentially similar to RET51 with deletion of residues 1073–1114, including Y1096). RET9(51C) is an allele where residues 1063–1072 of RET51 were replaced with residues 1063–1072 of RET9 (results in a receptor that is essentially similar to RET9 with C-terminus of RET51 including Y1096). (B) Hematoxylin-eosin-stained kidney sections of homozygous mice for the indicated knocked-in alleles at P0. Left column shows all the mutants in the context of ret51 isoform, and the right column in RET 9 context. Note wild-type forms of RET9 and RET51 have redundant functions in kidney development. Except for RET-Plcγ mutant RET51(Y1015F) individual abrogation of the key docking sites was dispensable. However, these same docking sites show dramatic kidney defects when the C-terminal Grb2 binding site is also deleted (mutations in RET9 context). Without Src and Grb2, there was a partially penetrant megaureter (see ); without Plcγ (individually or without Grb2 site) kidneys are small and show cysts and dysplasia; without Shc and Grb2 (RET9-Y1062F), only kidney rudiments or agenesis is seen. Thus direct Grb2 binding site provides some redundancy to Y981 and Y1062 docking sites in kidney development. In these studies, swapping the 10 residues following 1063aa between the two isoforms did not appear to influence kidney development suggesting that differences in the isoforms are not due to these regions. Adapted and modified from Jain et al.Citation42 with permission.
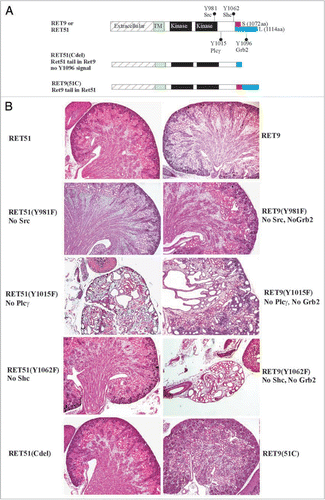
Figure 5 Complex CAKUT phenotype RET51(Y1015F) due to supernumerary ureters and decreased branching. (A) Histological and gross features of RET-Y1015F-PLCγ signaling mutant. The H&E-stained pictures at P0 show cystic hypodysplastic kidney (left), dilated ureters and possible distal stricture (middle) and failure of testes to separate from the urinary system (right). The gross photograph shows massive megaureter/hydroureter and barely any recognizable kidney parenchyma in 4 week old RET(Y1015F) mutant mice. (B) Whole mount Dolichos Biflorus Agglutinin-FITC (DBA-FITC) staining of E15.5 kidneys. Top, Normal honeycomb pattern of ureteric bud branching, single ureters entering the bladder (white dot), and separation of Wolffian duct (wd) from the ureter was evident in kidneys (K) of mice expressing WT, RET51 and RET51(Y1062F). In RET51(Y1015F) animals, note bilateral small kidneys with supernumerary UBs (arrows) that enter the mesenchyme but show decreased branching and failed to separate from the wd (white dashed line). The testes (T) were also abnormally positioned. Bottom, Imaging of the above kidneys by confocal microscopy clearly depicts the dramatic deficit in branching morphogenesis (arrowheads indicate branch points) in homozygous RET51(Y1015F) kidneys. Scale bar: 100 µm. Adapted and modified from Jain et al.Citation42 with permission.
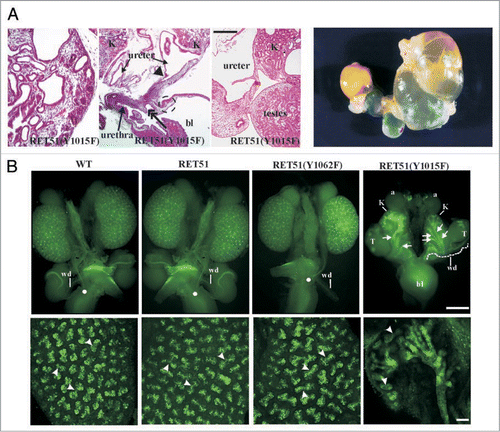
Figure 6 Genetic interactions between Ret and Sprouty1 (Spry1) are specific to kidney. (A) Loss of Sprouty1 rescues renal defects found in animals expressing Ret9 Y1062F. Whole mount of genito-urinary tracts from newborn mice of the indicated genotypes. Note that compared to individual Ret9-Y1062F or Spry1-/-, the double Ret9-Y1062F:Spry1-null mice have essentially normal appearing kidneys and ureters. Thus, both the agenesis and complex CAKUT phenotypes are rescued when these mutations are in the same background. (B) Lack of effect of Spry1 deficiency on the enteric innervation defects observed in RetRET9(Y1062F/Y1062F) mice, as revealed by whole mount acetylcholine esterase activity of digestive tracts from newborn mice. Thus, in the enteric nervous system Spry1 does not interact with Ret as it is not able to rescue Hirschsprung disease phenotype in Ret-Y1062F mutants. Adapted and modified from Rozen et al.Citation51 with permission. (C–F) Model summarizing Ret-Spry1 interactions in the kidney. In normal situations, Ret-activated cascades and Spry1 inhibition on Ret and other RTKs is in correct balance leading to normal kidneys (C). In absence of Ret-Y1062 signals, the main residue that determines uB outgrowth, kidneys don't develop; other RTKs cannot compensate as Spry1 is still inhibiting them (D). Without Spry1, Ret-MAPK inhibition is gone leading to perhaps enhanced Ret-Y1062-MAPK signaling, as is also from other RTKs that may have the potential to influence UB growth, thus leading to multiple ectopic UBs from the WD (E). In the absence of both Spry1 and Ret-Y1062, normal kidneys are observed because, other RTKs or other Ret-Tyr can potentially activate UB growth mechanisms in distal WD as repression from Spry1 is absent; ectopic UBs are not seen since RetY-1062 is the major signaling mechanism for UB growth in anterior WD.
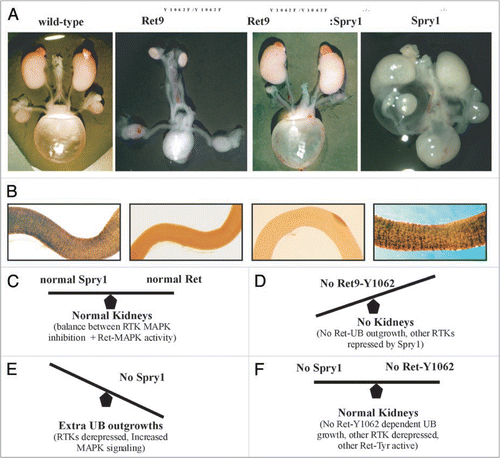
Figure 7 Many faces of Ret dysfunction in kidney. Schematic shows the major key Ret docking tyrosine and the preferred activated pathways. Also, depicted are the roles of these in the major processes of kidney development and examples of the phenotypes that are seen due to defects in these processes. The spectrum of phenotypes in Ret mutants are shown. Note that influence of other modifier genes such as Spry1 or Gdnf can give combination of intermediate phenotypes that are not represented here. Thus, alterations in Ret signaling pathways by changes in overall expression, or individual cascades. or through other genes encompasses majority if not all of CAKUT phenotypes. Parts of this figure are adapted from Jain et al.Citation40,Citation42 with permission.
Acknowledgements
I am highly grateful to members of my laboratory and collaborators whose work is presented in this manuscript. I thank Rajshekhar Chatterjee, Davis Keefe-Thomas and Masato Hoshi for their comments. I apologize to colleagues whose work could not be discussed or those whose work I may have inadvertently omitted. I am also grateful to George M. O'Brien Center for Kidney Disease Research (P30DK079333), NIDDK (DK081644, DK082531), NICHD (HD047396) and Childrens Discovery Institute at Washington University (MDII2009177) for supporting the work discussed here.
Note
Edited transcripts of research conferences sponsored by Organogenesis and the Washington University George M. O'Brien Center for Kidney Disease Research (P30 DK079333) are published in Organogenesis. These conferences cover organogenesis in all multicellular organisms including research into tissue engineering, artificial organs and organ substitutes and are participated in by faculty at Washington University School of Medicine, St. Louis Missouri, USA.
References
- Pope JC 4th, Brock JW 3rd, Adams MC, Stephens FD, Ichikawa I. How they begin and how they end: classic and new theories for the development and deterioration of congenital anomalies of the kidney and urinary tract, CAKUT. J Am Soc Nephrol 1999; 10:2018 - 2028
- Skinner MA, Safford SD, Reeves JG, Jackson ME, Freemerman AJ. Renal aplasia in humans is associated with RET mutations. Am J Hum Genet 2008; 82:344
- Saxen L. Organogenesis of the kidney 1987; Cambridge Cambridge University Press
- Costantini F, Shakya R. GDNF/Ret signaling and the development of the kidney. Bioessays 2006; 28:117 - 127
- Dressler GR. The cellular basis of kidney development. Annu Rev Cell Dev Biol 2006; 22:509 - 529
- Batourina E, Choi C, Paragas N, Bello N, Hensle T, Costantini FD, et al. Distal ureter morphogenesis depends on epithelial cell remodeling mediated by vitamin A and Ret. Nat Genet 2002; 32:109 - 115
- Takahashi M, Ritz J, Cooper GM. Activation of a novel human transforming gene, ret, by DNA rearrangement. Cell 1985; 42:581 - 588
- Ishizaka Y, Itoh F, Tahira T, Ikeda I, Sugimura T, Tucker J, et al. Human ret proto-oncogene mapped to chromosome 10q11.2. Oncogene 1989; 4:1519 - 1521
- Ishizaka Y, Ochiai M, Tahira T, Sugimura T, Nagao M. Activation of the ret-II oncogene without a sequence encoding a transmembrane domain and transforming activity of two ret-II oncogene products differing in carboxy-termini due to alternative splicing. Oncogene 1989; 4:789 - 794
- Boulay A, Breuleux M, Stephan C, Fux C, Brisken C, Fiche M, et al. The Ret receptor tyrosine kinase pathway functionally interacts with the ER{alpha} pathway in breast cancer. Cancer Res 2008; 68:374351
- Iwashita T, Kurokawa K, Qiao S, Murakami H, Asai N, Kawai K, et al. Functional analysis of RET with Hirschsprung mutations affecting its kinase domain. Gastroenterology 2001; 121:24 - 33
- Moore SW. The contribution of associated congenital anomalies in understanding Hirschsprung's disease. Pediatr Surg Int 2006; 22:305 - 315
- Prato AP, Musso M, Ceccherini I, Mattioli G, Giunta C, Ghiggeri GM, et al. Hirschsprung disease and congenital anomalies of the kidney and urinary tract (CAKUT): a novel syndromic association. Medicine 2009; 88:83 - 90
- Baloh RH, Enomoto H, Johnson EM Jr, Milbrandt J. The GDNF family ligands and receptors—implications for neural development. Curr Opin Neurobiol 2000; 10:103 - 110
- Arighi E, Borrello MG, Sariola H. RET tyrosine kinase signaling in development and cancer. Cytokine Growth Factor Rev 2005; 16:441 - 467
- Sariola H, Saarma M. Novel functions and signalling pathways for GDNF. J Cell Sci 2003; 116:3855 - 3862
- Golden JP, DeMaro JA, Osborne PA, Milbrandt J, Johnson EM Jr. Expression of neurturin, GDNF and GDNF family-receptor mRNA in the developing and mature mouse. Exp Neurol 1999; 158:504 - 528
- Schuchardt A, D'Agati V, Larsson-Blomberg L, Costantini F, Pachnis V. Defects in the kidney and enteric nervous system of mice lacking the tyrosine kinase receptor Ret. Nature 1994; 367:380 - 383
- Schuchardt A, D'Agati V, Pachnis V, Costantini F. Renal agenesis and hypodysplasia in ret-k-mutant mice result from defects in ureteric bud development. Development 1996; 122:1919 - 1929
- Sainio K, Suvanto P, Saarma M, Arumae U, Lindahl M, Davies JA, et al. Glial Cell-line derived neurotrophic factor is a morphogen for the ureteric bud epithelium. Development 1997; 20:4077 - 4087
- Heuckeroth RO, Enomoto H, Grider JR, Golden JP, Hanke JA, Jackman A, et al. Gene targeting reveals a critical role for neurturin in the development and maintenance of enteric, sensory and parasympathetic neurons. Neuron 1999; 22:253 - 263
- Yu T, Scully S, Yu Y, Fox GM, Jing S, Zhou R. Expression of GDNF family receptor components during development: implications in the mechanisms of interaction. J Neurosci 1998; 18:4684 - 4696
- Enomoto H, Hughes I, Golden J, Baloh RH, Yonemura S, Heuckeroth RO, et al. GFRalpha1 expression in cells lacking RET is dispensable for organogenesis and nerve regeneration. Neuron 2004; 44:623 - 636
- Paratcha G, Ledda F, Ibanez CF. The neural cell adhesion molecule NCAM is an alternative signaling receptor for GDNF family ligands. Cell 2003; 113:867 - 879
- Cacalano G, Farinas I, Wang LC, Hagler K, Forgie A, Moore M, et al. GFRalpha1 is an essential receptor component for GDNF in the developing nervous system and kidney. Neuron 1998; 21:53 - 62
- Enomoto H, Araki T, Jackman A, Heuckeroth RO, Snider WD, Johnson EM Jr, et al. GFR a1-deficient mice have deficits in the enteric nervous system and kidneys. Neuron 1998; 21:317 - 324
- Moore MW, Klein RD, Farinas I, Sauer H, Armanini M, Phillips H, et al. Renal and neuronal abnormalities in mice lacking GDNF. Nature 1996; 382:76 - 79
- Pichel JG, Shen L, Hui SZ, Granholm A-C, Drago J, Grinberg A, et al. Defects in enteric innervation and kidney development in mice lacking GDNF. Nature 1996; 382:73 - 76
- Sanchez MP, Silos-Santiago I, Frisen J, He B, Lira SA, Barbacid M. Renal agenesis and the absence of enteric neurons in mice lacking GDNF. Nature 1996; 382:70 - 73
- Tang MJ, Cai Y, Tsai SJ, Wang YK, Dressler GR. Ureteric bud outgrowth in response to RET activation is mediated by phosphatidylinositol 3-kinase. Dev Biol 2002; 243:128 - 136
- Tang MJ, Worley D, Sanicola M, Dressler GR. The RET-glial cell-derived neurotrophic factor (GDNF) pathway stimulates migration and chemoattraction of epithelial cells. J Cell Biol 1998; 142:1337 - 1345
- Watanabe T, Costantini F. Real-time analysis of ureteric bud branching morphogenesis in vitro. Dev Biol 2004; 271:98 - 108
- Shakya R, Watanabe T, Costantini F. The role of GDNF/Ret signaling in ureteric bud cell fate and branching morphogenesis. Dev Cell 2005; 8:65 - 74
- Chi X, Michos O, Shakya R, Riccio P, Enomoto H, Licht JD, et al. Ret-dependent cell rearrangements in the Wolffian duct epithelium initiate ureteric bud morphogenesis. Dev Cell 2009; 17:199 - 209
- Shakya R, Jho E-h, Kotka P, Wu Z, Kholodilov N, Burke R, et al. The role of GDNF in patterning the excretory system. Dev Biol 2005; 283:70 - 84
- Batourina E, Tsai S, Lambert S, Sprenkle P, Viana R, Dutta S, et al. Apoptosis induced by vitamin A signaling is crucial for connecting the ureters to the bladder. Nat Genet 2005; 37:1082 - 1089
- Viana R, Batourina E, Huang H, Dressler GR, Kobayashi A, Behringer RR, et al. The development of the bladder trigone, the center of the anti-reflux mechanism. Development 2007; 134:3763 - 3769
- Uetani N, Bertozzi K, Chagnon MJ, Hendriks W, Tremblay ML, Bouchard M. Maturation of ureter-bladder connection in mice is controlled by LAR family receptor protein tyrosine phosphatases. J Clin Invest 2009; 119:924 - 935
- de Graaff E, Srinivas S, Kilkenny C, D'Agati V, Mankoo BS, Costantini F, et al. Differential activities of the RET tyrosine kinase receptor isoforms during mammalian embryogenesis. Genes Dev 2001; 15:2433 - 2444
- Jain S, Naughton CK, Yang M, Strickland A, Vij K, Encinas M, et al. Mice expressing a dominant-negative Ret mutation phenocopy human Hirschsprung disease and delineate a direct role of Ret in spermatogenesis. Development 2004; 131:5503 - 5513
- Yu J, McMahon AP, Valerius MT. Recent genetic studies of mouse kidney development. Curr Opin Genet Dev 2004; 14:550 - 557
- Jain S, Encinas M, Johnson EM Jr, Milbrandt J. Critical and distinct roles for key RET tyrosine docking sites in renal development. Genes Dev 2006; 20:321 - 333
- Wong A, Bogni S, Kotka P, de Graaff E, D'Agati V, Costantini F, et al. Phosphotyrosine 1062 is critical for the in vivo activity of the Ret9 receptor tyrosine kinase isoform. Mol Cell Biol 2005; 25:9661 - 9673
- Jijiwa M, Fukuda T, Kawai K, Nakamura A, Kurokawa K, Murakumo Y, et al. A targeting mutation of tyrosine 1062 in Ret causes a marked decrease of enteric neurons and renal hypoplasia. Mol Cell Biol 2004; 24:8026 - 8036
- Chi L, Zhang S, Lin Y, Prunskaite-Hyyrylainen R, Vuolteenaho R, Itaranta P, et al. Sprouty proteins regulate ureteric branching by coordinating reciprocal epithelial Wnt11, mesenchymal Gdnf and stromal Fgf7 signalling during kidney development. Development 2004; 131:3345 - 3356
- Basson MA, Akbulut S, Watson-Johnson J, Simon R, Carroll TJ, Shakya R, et al. Sprouty1 is a critical regulator of GDNF/RET-mediated kidney induction. Dev Cell 2005; 8:229 - 239
- Grieshammer U, Le M, Plump AS, Wang F, Tessier-Lavigne M, Martin GR. SLIT2-mediated ROBO2 signaling restricts kidney induction to a single site. Dev Cell 2004; 6:709 - 717
- Kume T, Deng K, Hogan BL. Murine forkhead/winged helix genes Foxc1 (Mf1) and Foxc2 (Mfh1) are required for the early organogenesis of the kidney and urinary tract. Development 2000; 127:1387 - 1395
- Basson MA, Watson-Johnson J, Shakya R, Akbulut S, Hyink D, Costantini FD, et al. Branching morphogenesis of the ureteric epithelium during kidney development is coordinated by the opposing functions of GDNF and Sprouty1. Dev Biol 2006; 299:466 - 477
- Mason JM, Morrison DJ, Basson MA, Licht JD. Sprouty proteins: multifaceted negative-feedback regulators of receptor tyrosine kinase signaling. Trends Cell Biol 2006; 16:45 - 54
- Rozen EJ, Schmidt H, Dolcet X, Basson MA, Jain S, Encinas M. Loss of Sprouty1 rescues renal agenesis caused by Ret mutation. J Am Soc Nephrol 2009; 20:255 - 259
- Celli G, LaRochelle WJ, Mackem S, Sharp R, Merlino G. Soluble dominant-negative receptor uncovers essential roles for fibroblast growth factors in multi-organ induction and patterning. EMBO J 1998; 17:1642 - 1655
- Maeshima A, Sakurai H, Choi Y, Kitamura S, Vaughn DA, Tee JB, et al. Glial cell derived neurotrophic factor independent ureteric bud outgrowth from the Wolffian duct. J Am Soc Nephrol 2007; 18:3147 - 3155
- Lu W, van Eerde AM, Fan X, Quintero-Rivera F, Kulkarni S, Ferguson H, et al. Disruption of ROBO2 is associated with urinary tract anomalies and confers risk of vesicoureteral reflux. Am J Hum Genet 2007; 80:616 - 632
- Weber S, Moriniere V, Knuppel T, Charbit M, Dusek J, Ghiggeri GM, et al. Prevalence of mutations in renal developmental genes in children with renal hypodysplasia: results of the ESCAPE study. J Am Soc Nephrol 2006; 17:2864 - 2870
- Tabatabaeifar M, Schlingmann KP, Litwin M, Emre S, Bakkaloglu A, Mehls O, et al. Functional analysis of BMP4 mutations identified in pediatric CAKUT patients. Pediatr Nephrol 2009; In press
- Vats KR, Ishwad C, Singla I, Vats A, Ferrell R, Ellis D, et al. A locus for renal malformations including vesico-ureteric reflux on chromosome 13q33-4. J Am Soc Nephrol 2006; 17:1158 - 1167
- Nakano T, Niimura F, Hohenfellner K, Miyakita E, Ichikawa I. Screening for mutations in BMP4 and FOXC1 genes in congenital anomalies of the kidney and urinary tract in humans. Tokai J Exp Clin Med 2003; 28:121 - 126
- Kim HJ, Bar-Sagi D. Modulation of signalling by Sprouty: a developing story. Nat Rev Mol Cell Biol 2004; 5:441 - 450
- Madhani HD. Accounting for specificity in receptor tyrosine kinase signaling. Cell 2001; 106:9 - 11