Abstract
Urinary tract obstruction leads to obstructive nephropathy, which in turn, frequently results in renal failure. Congenital urinary tract obstruction can be traced back to errors during the organogenesis of the urinary system. A fundamental understanding of the causes of urinary tract obstruction and the developmental processes involved are critical for improving the diagnostic and therapeutic strategies for this disease. A number of laboratories, including ours, have been using genetically engineered and spontaneously occurring mouse models to study the primary causes and the pathogenesis of urinary tract obstruction. These studies have shown that urinary tract obstruction is a very heterogeneous disease that can be caused by a diverse set of factors targeting multiple levels of the urinary system. Accumulating evidence also indicates that the development of the urinary tract requires the integration of progenitor cells of diverse embryonic origins, leading to the formation of multiple junctions prone to developmental errors. In addition, the high sensitivity of the pyeloureteral peristaltic machinery to disturbance affecting the structural or functional integrity of its components also contributes to the high incidence rate of urinary tract obstruction.
Acknowledgements
The author wishes to thank Drs. Helen Liapis, and Qiusha Guo for critical reading of the manuscript. F.C. has been supported in part by a NIH grant (DK067386) and the George M. O'Brien Washington University Center for Kidney Disease Research (NIHP30DK079333).
Note
Edited transcripts of research conferences sponsored by Organogenesis and the Washington University George M. O'Brien Center for Kidney Disease Research (P30 DK079333) are published in Organogenesis. These conferences cover organogenesis in all multicellular organisms including research into tissue engineering, artificial organs and organ substitutes and are participated by faculty at Washington University School of Medicine, St. Louis, Missouri.
Figures and Tables
Figure 1 The Calcineurin-NFAT signaling pathway. Calcineurin is a calcium-dependent serine-threonine phosphatase. Calcineurin has a catalytic subunit “A” and a regulatory subunit “B”. Both of these subunits are indispensible for calcineurin activity. The activity of calcineurin can be inhibited by a number of intrinsic and pharmacological inhibitors, such as Cyclosporin A (CsA) and FK506. The increase in cytoplasmic calcium level can be triggered by many factors, including the activation of a diverse set of cell surface receptors, ion channels and even gap junctions. Intracellular calcium increase leads to the activation of calcineurin. Activated calcineurin dephosphorylates its substrates, including the NFATc transcription factors. The dephosphorylated NFATc proteins translocate from the cytoplasm to the nucleus to regulate the transcription of their target genes. The regulation of transcription by NFATc proteins requires nuclear partners (NFATn). These nuclear partners can be different for different target genes and in different cells. These nuclear partners are usually activated by a different set of receptors on the cell surface.
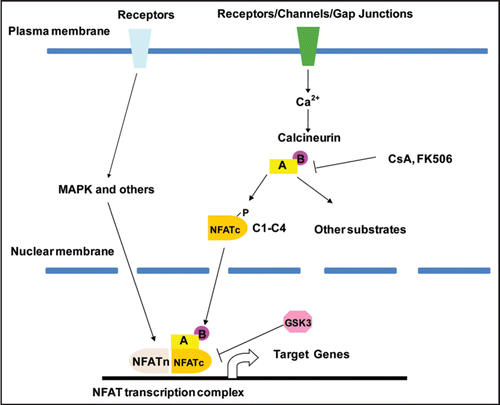
Figure 2 Inactivation of Cnb1 in the metanephric and ureteric mesenchyme causes congenital obstructive nephropathy. (A–E) are samples from P1 Pax3-CreT/+, ROSAT/+ mice. In the kidney proper, LacZ expression (blue, reflecting the Cre expression) is evident in the glomeruli and tubules that are MM-derived but not in the collecting duct system originated from the UB (A). The black arrow in (B) points to the developing glomeruli. The open arrow points to one of the UB branches. In the developing renal pelvic region, the connective tissues, the smooth muscle layers (SM) and the adventitia (AD) of the urinary tract express LacZ while the UB-derived urothelium (UT) remains LacZ-negative (C–E). U: ureter; PP: Papilla. The SM layers in the developing renal pelvic wall are illustrated by αSMA staining on a wild-type newborn sample of the same area (D). LacZ is also selectively expressed in the SM layers in the ureter but not in the stratified transitional epithelium (E). Mutants (Pax3-CreT/+, Cnb1lox/lox) have severe hydronephrosis and erosion of the kidney parenchyma (G and I compared with F and H. Samples G and H were collected at P12). Modified from and from ref. Citation26 with permission.
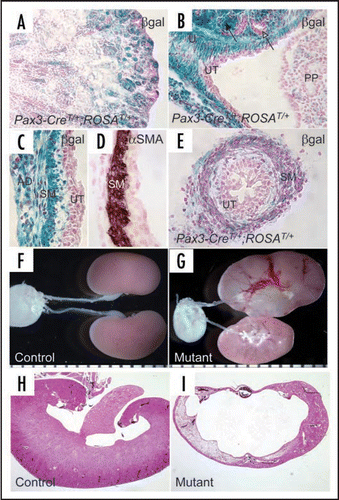
Figure 3 Inactivation of Cnb1 in the metanephric and ureteric mesenchyme disrupts the development of the pyeloureteral peristaltic machinery. (A and B) are hemi-sected control (A) and mutant (B) kidneys. The white arrows point to the UPJ. (C and D) are sections of the developing renal pelvic wall of control (C) and mutant (D) immunostained with a Ki67 antibody. The controls have significantly more proliferating mesenchymal cells along the developing renal pelvic wall. PP: papilla. M: mesenchymal derivatives. UT: urothelium. Modified from and from ref. Citation26 with permission.
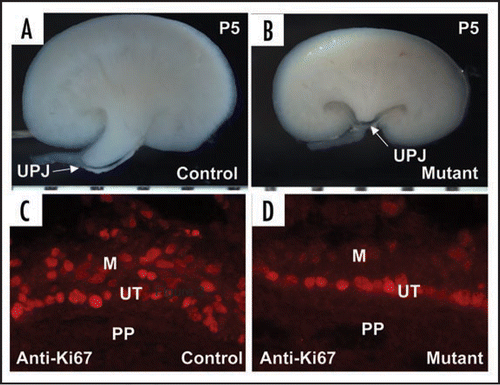
Figure 4 Genetic mapping of the cph mutants with congenital obstructive nephropathy. (A–C) The mutants have unilateral or bilateral hydronephrosis and hydroureter (arrow). (D) The BUN level in the mutants (P5–P16) is dramatically increased. (E and F) Kidney sections from P14 control and mutant littermates stained with H&E. (G) Physical map of relevant portions of mouse chromosome 15. Our genetic mapping efforts locate the cph locus to the chromosomal interval of about 0.7 Mbp, defined by the microsatellite markers C15LD6 and C15LD5. The black triangle indicates the chromosomal location of Aqp2. Modified from and from ref. Citation43 with permission.
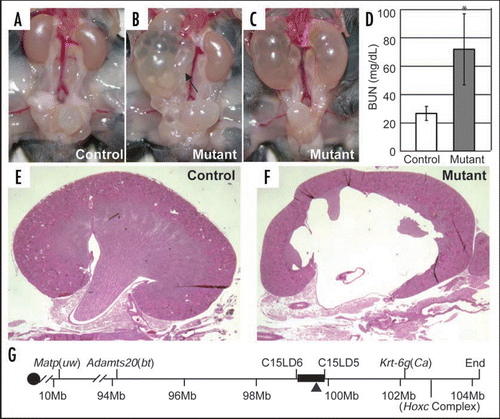
Figure 5 A mutation in Aqp2 disrupting its trafficking causes urine concentration defect and obstructive nephropathy. (A and B) Immunostaining of the collecting ducts in the outer medulla shows apical accumulation of Aqp2 in the controls (A) but a diffuse staining with no apical accumulation in the mutant (B). (C) Sequence of the fourth exon of Aqp2 reveals the C-T substitution at nucleotide 767 in the homozygous mutants, while the heterozygotes have both C and T represented at position 767. This substitution results in a Ser to Leu change at amino acid 256 in the cytoplasmic tail of the Aqp2 protein. +, wild-type allele; c, cph mutant allele. Modified from from ref. Citation43 with permission.
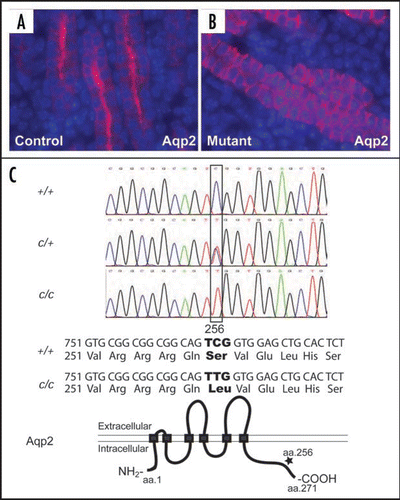
Figure 6 Urinary tract development requires the integration of diverse progenitor cell populations and the formation of multiple junctions. Nephron components, excluding the collecting ducts, are derived from the MM within the intermediate mesoderm. The collecting duct and the ureteric epithelium are derived from the UB. The tailbud-derived mesenchyme gives rise to the UM that in turn produces the smooth muscles and other mesenchymal derivatives along the ureter and along the ureter. While the vesical (bladder) epithelium is from the hindgut endoderm, the vesical mesenchyme appears to arise from tailbud-derived mesenchyme. The lineage origin of the urethral mesenchyme is not entirely clear, but the urethral epithelium is also derived from the same hindgut endoderm that gives rise to the vesical epithelium. The origin of the renal capsule is less clear but appears to be distinct from the other structures within the urinary system. The use of different colors is for the distinction of tissues with different embryonic origins. The different colors used for the UM and vesical mesenchyme (both from the tailbud-derived mesenchyme) reflect the fact that they develop as separate structures before being joint together later in development. The entire urinary tract is also highly innervated. The integration of these diverse progenitor populations also involves the formation of junctional complexes, especially at the UPJ and UVJ. Any disruption of the regulation of the integration could lead to urinary defects, including urinary tract obstruction.
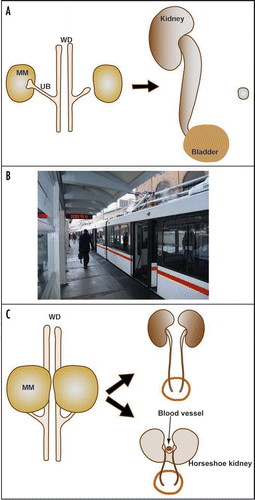
Figure 7 Quantitative differences can be translated into qualitatively distinct outcomes in urinary tract development, contributing the phenotypic variations. (A) In a hypothetical scenario, a genetic defect affects UB budding to a degree that the UB barely reaches the MM just in time to prevent its apoptosis. Other problems associated with this mutation then lead to the development of urinary tract obstruction. Any additional disturbance may delay the UB outgrowth further, causing the failure of the UB to reach MM before its degeneration and leading to renal agenesis. This is similar to a scenario (depicted in B) where you are running late to catch a train. You may be lucky to get on it at the last second, or your taxi to the station may hit one red light too many and you miss it all together. In another hypothetical example (C), abnormal intermediate mesoderm patterning causes the MM from both sides to position closer to the midline than normal. The left and right kidneys may develop relatively normally despite their unusual positions. However, the close proximity of the MM from both sides sets up an unstable state that any slight variation may cause MM fusion and the occurrence of horseshoe kidneys that tend to be obstructed. Therefore, very slight and random variations may separate a rather healthy state and a potentially very morbid state.