Abstract
The key pathogenic event in prion disease involves misfolding and aggregation of the cellular prion protein (PrP). Beyond this fundamental observation, the mechanism by which PrP misfolding in neurons leads to injury and death remains enigmatic. Prion toxicity may come about by perverting the normal function of PrP. If so, understanding the normal function of PrP may help to elucidate the molecular mechansim of prion disease. Ablation of the Prnp gene, which encodes PrP, was instrumental for determining that the continuous production of PrP is essential for replicating prion infectivity. Since the structure of PrP has not provided any hints to its possible function, and there is no obvious phenotype in PrP KO mice, studies of PrP function have often relied on intuition and serendipity. Here, we enumerate the multitude of phenotypes described in PrP deficient mice, many of which manifest themselves only upon physiological challenge. We discuss the pleiotropic phenotypes of PrP deficient mice in relation to the possible normal function of PrP. The critical question remains open: which of these phenotypes are primary effects of PrP deletion and what do they tell us about the function of PrP?
Introduction
The prion protein (PrPC) is a conserved glycoprotein tethered to cell membranes by a glycosylphosphatidylinositol (GPI) anchor.Citation1 PrPC denotes “cellular” or “normal” PrP to differentiate it from PrPSc for “scrapie” or disease associated isoform of PrPC. PrPC is expressed in many tissues, most abundantly in brain, heart, muscle, and also in select lymphoid and myeloid cells.Citation2 The role of PrPSc in the pathogenesis of the transmissible spongiform encephalopathies (TSE), the prion diseases, has been intensively studied.Citation1–Citation5 Conversely, much less attention has been focused on the role of PrPC in normal physiology.Citation6 Of note, normal function studies of proteins associated with other neurodegenerative diseases, such as amyloid precursor protein and the secretases for Alzheimer's disease,Citation7 α-synuclein for Parkinson's disease,Citation8 and huntingtin for Huntington's disease,Citation9 are helping to provide deeper insights into the pathophysiology of these diseases. Analogously, a clearer understanding of the function of PrPC in homeostasis may provide valuable insights into the molecular pathways of prion pathogenesis. However, the extent of overlap between understanding the pathogenic dysfunction of PrPSc in prion diseases and the normal function of PrPC in cell physiology remains to be determined (depicted in ).
Many approaches have been utilized to understand the physiological function of PrPC, including but not limited to the identification of multiple interaction partners, human genetic studies of the Prnp (prion protein gene) locus, ectopic and overexpression of PrPC in a variety of cell types and organisms, and finally deletion or ‘knockout’ (KO) studies in the mouse,Citation10 cow,Citation11 and even goat,Citation12 providing additional exciting tools for understanding aspects of PrPC physiology that may not be addressable in mice (). The search for protein interaction partners of PrPC, by a variety of methods, has led to interesting candidates but functional demonstration of the importance of these interactions is still missing.Citation2,Citation13 Furthermore, overexpression and ectopic expression studies of PrPC, or expression of mammalian PrPC in lower organisms is yet to reveal an irrefutable function for PrPC. Another approach includes large scale genetic association studies to look for PRNP mutations or polymorphisms associated with human genetic disorders. Some interesting genetic associations with Alzheimer's disease susceptibility have been found but lack consensus in the field.Citation14–Citation16 Polymorphisms in PrPC have been associated with rare forms of cortical malformation,Citation17 differences in surgical outcome for a form of epilepsy,Citation18 and even learning and memory.Citation19
A large pool of PRNP mutations have been identified in humans, but all of these are associated with familial prion diseases, or appear to represent harmless polymorphisms.Citation20 Perhaps there are heretofore undiscovered humans with small chromosomal deletions that encompass the PRNP locus, or even mutations that render humans haploinsufficient or even null for PrP. Such human patients may tell us volumes about the function of PrPC and perhaps that it is dispensable for normal life as is the case for miceCitation10 and cows.Citation11
Because PrPC is so conserved among mammals, there was great expectation that ablation of Prnp in the mouse would reveal a normal function for this enigmatic gene.Citation21 Since the PrP KO mice have no overt phenotype,Citation10,Citation22 it was clear that PrPC is not essential for the survival of the laboratory mouse. However, genetic compensation and developmental plasticity may mask the phenotype of PrPC deficient mice and thus, it may take an appropriate challenge to reveal any phenotype. Although the original reports on the Zurich and Edinburgh PrP KO mice (two different targeting strategies to delete PrP, named after the city where the experiments took place) reported “no phenotype”, many subsequent studies have revealed that this KO mouse has an abundance of phenotypes, some of which have been contested and many of which are subtle (). This review summarizes the recent research in determining the normal function of PrPC by utilizing the PrP KO mouse. Although many claims to PrPC function have been generated from the study of cultured cells,Citation3,Citation23 we will mostly confine our discussion to studies utilizing mice: the PrP KO, the deletion of the PrPC homolog doppel (Dpl), and overexpression transgenics of PrPC and Dpl. Prnp is one of the most frequently knocked out mammalian genes, and a plethora of PrP-deficient mice have been generated with a wealth of strategies. Detailed reviews of the construction of the various available PrP KO mice exists.Citation24,Citation25 The neurodegeneration caused by the ectopic expression of Dpl in the Nagasaki and Rcm PrP KO (generated with a different gene targeting strategy than the Zurich and Edinburgh PrP KOs) has been reviewed in detail,Citation24,Citation25 and will be discussed only in so far as PrPC and Dpl function(s) are concerned.
The Caveats of A Phenotype
Given the abundance of phenotypes attributed to the deletion of PrP, we will first discuss several technical concerns relating to KO studies and potential caveats of phenotypes in PrP KO mice. The advent of “gene-targeting”, or the specific deletion/replacement of chromosomal segments, in mice has revolutionized functional studies of mammalian genes in vivo. However, technical aspects of generating gene-targeted mice from embryonic stem cells can create a potential caveat to interpreting phenotypic data. This caveat arises because often the resultant mice utilized for functional studies are maintained on “mixed” genetic backgrounds, a random mixture of alleles from embryonic stem cells (often a 129 sub-strain) and the parental line (C57Bl/6, Balb/c, or others). Three concerns arise when working on mice of a mixed genetic background, which encompass most studies of PrP KO mice: 1) increases in the phenotypic variability and thus a higher chance of spurious results 2) the heterozygosity at many loci increases the noise of most measurements, potentially obscuring subtle phenotypes, and 3) the possibility that alleles linked to the deleted gene (which is continually selected for by investigators during breeding) are actually responsible for the phenotype in question.Citation26 Another point to consider is that depending on how the investigator maintains the KO line (either by inbreeding separate populations of PrP-/- and PrP+/+, or by intercrossing PrP+/- mice) we also have to contend with genetic drift, that is the tendency for certain alleles to become “fixed” (100% frequency) in populations simply by chance.
The availability of the fully-sequenced mouse genome, in concert with physical maps of polymorphic short-tandem repeats, would allow for eliminating the confounding genetic factors enumerated above. For example, the “speed congenic” technologies which are being offered commercially—but can be enacted in-house by real-time PCR and sequencing—enable selective breeding of mice that are genetically homogeneous at any given chromosomal region. However, almost none of the phenotypes described in the following pages have been ascertained using such genetic controls—and this represents a major caveat in the interpretation of phenotypic data. Of the various PrP KOs that have been generated,Citation25 only the Edinburgh KO has been maintained on a pure 129/Ola backgroundCitation22 and this strain background is notoriously difficult to work with because of poor breeding and several other abnormalities.Citation26 Backcrossing PrP KOs onto a pure genetic background helps to eliminate this concern. However, despite intensive backcrossing, regions on the chromosome adjacent to the Prnp locus are carried along through all the generations of backcrossing, such that even after 12 generations of backcross the KO allele would be flanked by about 1% of the ES cell genome.Citation26 As a consequence, it is difficult to conclude with certainty that a phenotype is due to the genetic background of the mouse or the deleted gene in question. Another point worth considering is that on pure backgrounds it is possible to obtain a phenotype that is a combination of the deletion in question (i.e., PrP KO) and a specific allele(s) of the background (i.e., a C57Bl/6 allele of a gene). Such a KO and strain background synergistic phenotype would be difficult to replicate unless other investigators are using the same mice.
Rescue experiments, whereby a transgene is reintroduced into the KO to rescue a phenotype, are time-consuming and are not “fool-proof ”, but represent one approach toward eliminating an effect of a linked allele or a chance observation in a mixed genetic background. On the other hand, such reliance on rescue experiments led to the erroneous assignment of pathological phenotypes to the deficiency of PrP (in Nagasaki PrP KOs),Citation27 whereas the phenotype was in reality brought about by the overexpression of Dpl in a PrP KO background.Citation25
A separate concern from genetic background arises from intensive and multi-faceted hunting for phenotypes. Since all biological measurements, but particularly those made on animals, are associated with intrinsic variability, we expect to find some spurious phenotypes just by chance. For example, if the null hypothesis is that PrP KOs are not different from wild-type with a conventional threshold of significance of p < 0.05, we expect that if 100 labs study the PrP KO that as many as five may find spurious phenotypes. This is an example of the “multiple hypothesis testing problem” and is an inescapable reality of biological research. Independent confirmation of findings in more than one type of the PrP KO mouse, for example the Edinburgh or Zurich PrP KOs, or by more than one laboratory will be important for building confidence in PrP KO phenotypes.
The Clearest of PrP Phenotypes: PrP Knockout Mice are Resistant to Prion Infection and Cannot Replicate Prions
To date, the clearest phenotype of the PrP KO is resistance to infection with prions.Citation28,Citation29 This experiment—originally designed to disprove the prion hypothesisCitation21—solidified its central tenet: the requirement of a host protein for prion replication. Further variations of this experiment have taught us that PrPC expression is required for prion-induced toxicity.Citation30 Neurografting brain tissue from wild-type mice into PrP KO brains revealed that Prnp+/+ tissue grafts replicated prions with accompanying damage to neurons while nearby PrP-deficient tissue was unharmed.Citation31 Thus, PrPC on neuronal cells is required for prion propagation associated toxicity. This evidence supports our conjecture that deciphering the normal function or signaling pathway through which PrPC operates will help illuminate the devastating sequence of events in prion disease.
The PrP KO mouse enabled another important series of studies that defined some of the sequence elements required for PrPSc to retain infectivity by expressing truncated PrP transgenes on the KO background and infecting these mice with prions. This approach revealed that the octapeptide repeats () were not necessary for prion replication or toxicity, but indicated that they may be required for the rampant spongiosis and production of high titers of prion infectivity normally associated with prion diseases.Citation32,Citation33
PrP in Sleep Regulation
Sleep disturbances and altered circadian rhythms were the first documented phenotypes in PrP KOs,Citation34 other than the resistance to prion infection which is technically a lack of a phenotype.Citation28 PrPC might be involved in regulating sleep, as certain mutations in PRNP cause a prion disease known as fatal familial insomnia.Citation35 This disease eventually results in broadly disseminated neurodegeneration but one key symptom is a nearly complete inability to sleep. Sleep deficits are also a documented feature in human Creutzfeldt-Jakob disease of sporadic origin.Citation36 Tobler and colleagues found that during a normal light/dark cycle PrP KOs had similar patterns of running wheel activity as controls. However, in constant darkness, wild-type mice display a shorter circadian period (as is customary for wild-type mice without circadian cues) whereas PrP KOs remarkably maintain a normal period as if still “entrained” by light.Citation34 This finding was shown in both the Edinburgh and Zurich PrP KOs. Further studies by Tobler and colleagues revealed that PrP KOs have more fragmented sleep episodes than do controls, leading the authors to conclude that PrPC plays a role in promoting sleep continuity.Citation37 The fact that PrPC alters sleep and that PrPSc production also leads to sleep abnormalities supports our hypothesis that understanding PrPC function will help to understand prion disease. But in nearly a decade since this phenotype was documented there is still no clarity as to how PrPC regulates sleep at a molecular level.
A Role for PrP in Oxidative Stress: Copper Binding, SOD-Activity and Mitochondria
A considerable amount of work has focused on the copper (Cu) binding and potential anti-oxidant function of PrPC. The genesis of this work is the observation that recombinant PrPC binds to Cu and that copper levels were diminished in brains of PrP KOs.Citation38 The ability of PrPC to bind Cu has been well supported but alteration in Cu content in PrP KO brains is controversial.Citation39 A study by Wong et al. suggests that PrPC is involved in defense against oxidative damage.Citation40 They observed higher levels of oxidized proteins and lipids in the brains of Edinburgh PrP KOs.Citation40 A similar situation was observed in Zurich PrP KOs by Klamt and colleagues, who further found that superoxide dismutase (SOD) activity was significantly decreased in the brain and muscle of PrP KOs.Citation41 On the other hand, Waggoner et al. could not detect differences in enzymatic activity of Cu-Zn superoxide dismutase.Citation39 This may be due to differences in experimental conditions.Citation42 However, mouse genetic experiments argue against a SOD-like activity for PrPC in vivo.Citation43 We reasoned that if PrPC has a SOD activity, then deficiency in both PrPC and SOD1 will result in diminished SOD activity compared to deficiency in SOD1 only, and conversely PrPC overexpression in a SOD1 KO or WT background will result in increased SOD activity. However, these genetically defined crosses did not reveal any alterations in SOD activity with respect to PrPC deletion.Citation43
One intriguing phenotype at the cellular level is the reduction in number of mitochondria per cell in the CA1 region of the hippocampus and in the myocardium of Edinburgh PrP KOs.Citation44 The mice used in this study were inbred 129/Ola strain, which reduces the likelihood of genetic background artifacts but could also result in strain specific effect of a gene deletion (discussed above). This study was stimulated by the identification of differentially expressed genes in the PrP KO, revealing only three genes that were differentially expressed-all of which were involved in mitochondrial biogenesis and physiology.Citation44 Also, mitochondria having morphological abnormalities were more abundant in the PrP KO (Edinburgh).Citation44 It is noteworthy that we identified swollen mitochondria in a transgenic model of prion disease, where PrP is mislocalized to the cytosol, another possible connection between PrPC in normal physiology and disease.Citation45 Lobao-Soares and colleagues sought to extend the observation of abnormal mitochondria in PrP KOs to a functional level by measuring mitochondrial respiration in several regions of PrP KO brains, but found similar levels between PrP KOs and controls in all regions examined.Citation46 However, it should be noted that isolated mitochondria were utilized in this study and it is possible that small numbers of abnormal mitochondria may not be detected by their assays. Further studies will be required to clarify the role of PrPC—if any—in oxidative stress and mitochondrial physiology.
The Role of PrP in the Immune System, Phagocytosis and as a Microbial Receptor
Recent reports have suggested a role for PrPC in cellular internalization pathways, perhaps a function that has been co-opted by microbes. Work from Rafael Linden and colleagues add to the growing list of phenotypes for the PrP KO. Prnp null macrophages displayed increased rates of phagocytosis in vitro and in vivo, leading to the conclusion that under physiological conditions PrPC negatively regulates phagocytosis.Citation47 This is extremely surprising since the expression levels of PrPC in macrophages are typically below the limits of detection (Christina Sigurdson and A. Aguzzi, unpublished data). Studies of microbial pathogenesis in PrP KOs may be connected with a role for PrPC in phagocytosis or cytokine production. Watarai and colleagues have discovered that the PrP KO is more resistant to infection with the bacterial pathogen Brucella abortus.Citation48,Citation49 Localization and biochemical experiments pinpointed bacterial heat shock protein 60 as an interaction partner with PrPC on the cell surface.Citation48 It would be interesting to examine the consequences of PrPC overexpression and B. abortus infection. These results were not confirmed in a different laboratory in studies using B. suis.Citation50
Virologists have made use of the PrP KO as well, revealing yet another phenotype likely relating to cellular internalization pathways. In this study, Thackray and Bujdoso demonstrate that PrP KOs are refractory to infection with a neurotropic herpes simplex virus whereas PrP overexpression transgenics (Tga20) were highly susceptible to infection.Citation51 Studies of viral titers and maturation markers suggest that viral replication is retarded in PrP KOs in favor of establishing latency. Intriguingly, the viral infection induced neuronal cell death much more dramatically in PrP overexpression transgenics, connecting PrPC to cell survival pathways (discussed below). Several of these findings were confirmed in a follow-up study by the same group.Citation52
Recent work suggests that PrPC may play a role in the immunological synapse. Ballerini et al., demonstrated that PrPC is important in an interaction between T cells and dendritic cell.Citation53 PrPC was dispensable on T cells for this interaction but PrPC on dendritic cells was important in stimulating T cells in vitro and in vivo assay.Citation53 Another study reports that αβ T cells are greatly diminished in tga20 PrP overexpression transgenic mice, and these mice also display an atrophy of the thymus.Citation54 However, this phenotype may represent an insertional mutagenesis artifact since tga19 transgenic mice derived from the same construct do not show these anomalies (AA, unpublished data). One study has attempted to link copper uptake and interleukin expression in T cells, finding a slight delay in interleukin-2 expression in PrP KO T cells.Citation55 To summarize, PrPC may be important for host-pathogen interactions, immune synapses and T cell homeostasis, but further studies will be needed to decipher the role of PrPC in the immune system.
Neuronal Excitability
The high level of PrPC expression in neuronal cells led to an interest in detecting electrophysiological defects in the PrP KO. This topic is no less controversial than any other we have discussed in this review, but the weight of the evidence clearly lies on the side of altered neuronal excitability in PrP KO neurons. Electrophysiological studies of PrP KOs were first under-taken by John Collinge and John Jefferys, who found that CA1 neurons in Zurich PrP KOs had faster after-hyperpolarization currents and were impaired in long term potentiation (LTP).Citation56 Jean Manson and colleagues had similar findings in purebred Edinburgh PrP KOs.Citation57 In addition it was shown that both wild-type and a familial mutant human PrPC were capable of rescuing this electrophysiological phenotype when expressed as transgenes in the PrP KO background.Citation58,Citation59 Soon after these findings were reported opposing reports surfaced. Herms et al. examined synaptic transmission in Purkinje cells of Zurich PrP KOs but did not detect any differences from controls.Citation60 Another group found no differences between Zurich PrP KOs and controls in the CA1 region of the hippocampus.Citation61 Over the ensuing years there have been several other attempts to clarify the electrophysiological phenotype (or lack thereof ) in PrP KOs.Citation62–Citation64 However, only one thing is clear—detection of the electrophysiological phenotype depends on which line of mice is being used, who is investigating, and the age of the PrP KOs being used.Citation65 It is worth noting that authors on two of the papers reporting “no phenotype”Citation60,Citation61 have reversed their position in later studies.Citation66,Citation67 Finally, the post natal neuronal-specific KO of PrPC showed a reduction of after-hyperpolarization potentials in neurons in CA1,Citation68 an identical phenotype to what had been originally reported by Collinge and colleagues.Citation56
The neuronal excitability phenotypes may relate to one of the strongest phenotypes of PrP KOs, which presents under the challenge of seizure inducing drugs. Zurich PrP KOs are much more susceptible to repeated doses of pentylene tetrazol and kainic acid, both of which induce seizures.Citation69 Approximately 50% of PrP KOs died from a single administration of kainic acid while 100% of control animals survived.Citation69 This result has been confirmed independently by Rangel, et al., who also note increased neuronal cell death in PrP KOs injected with kainic acid.Citation70 The increased seizure sensitivity may be due to higher levels of ectonucleotidase activity which destroys adenosine, an endogenous anticonvulsant agent, in PrP KOs.Citation71,Citation72 Finally, it is also worth noting that a defect in neuronal architecture of the hippocampus in Zurich PrP deficient mice has been reported and may be relevant to several of the findings discussed above.Citation73 Timm stained sections of the hippocampus from PrP KO had more sprouting of axons than did controls in the granule cell layer of the dentate gyrus and the infrapyramidal region of CA3 region.Citation73 This is said to resemble the mossy fiber collateral and terminal sprouting seen in certain human epilepsies.
Behavioral Phenotypes: Is PrP Involved in Learning and Memory?
If they bear any relevance to real life, the electrophysiological defects described above for the PrPC null neurons might manifest in the behavior of the PrP KO mouse. The abundant expression of PrPC in regions important in learning and memory, such as the hippocampus, has lead to a series of behavioral studies aimed at detecting abnormalities in PrP KOs. Initial studies by Charles Weissmann, Hans Peter Lipp, and colleagues did not detect any phenotype of Zurich PrP KOs in a long-term study using maze tests.Citation10,Citation74 Further, a study by Roesler et al. failed to detect any abnormalities in anxiety nor inhibitory avoidance learning in PrP KOs.Citation75 Cognitive defects have been detected in PrP KOs by Criado and colleagues who found that spatial learning was defective in PrP KOs.Citation62 This phenotype was PrP-dependent as it was rescued by crossing PrP KO mice with a transgene driving expression of hamster PrPC under a neuron-specific promoter.Citation62 Another study describes PrP KOs as having normal short- and long-term memory at three months of age but impairments by 9 months of age.Citation76 Aged PrP KOs also showed less exploratory activity in an open field.Citation76 A follow-up study suggested that the interaction of PrPC and laminin may be key to memory consolidation in rats,Citation77 although there is no clarity about which molecular events might be triggered by the binding of PrPC to laminin. Finally, a study in humans suggests that the M129V polymorphism in PrPC, which influences susceptibility to prion infection in humans-may be involved in learning and memory.Citation19
In an attempt to reveal a behavioral phenotype, investigators have challenged PrP KOs in various ways during phenotypic testing. Coitinho et al. dosed PrP KO mice with various psychotropic drugs and interestingly, PrP KOs show a decreased response to the psychotropic drug MK-801, which normally causes increased motor activity.Citation78 Amphetamine and caffeine induced hyper-locomotion to an equal extent in PrP KOs and controls.Citation78 Nico et al. subjected Zurich PrP KOs to acute stress by foot shock or swimming trial and found that PrP KOs showed less anxiety than controls after these treatments.Citation79 In non-stress conditions, PrP KOs appeared identical to controls.Citation79 Another study notes a very subtle increase in locomotor activity in PrP KOs in an open field test.Citation75 This increased locomotor activity has not been observed using extensively backcrossed C57Bl/6 PrP KOs (both Edinburgh and Zurich) in the home cage using high resolution techniques recently used to study prion disease in detail,Citation80 however, our testing conditions are not equivalent to an open field test (ADS and SL, unpublished results).
Diverse Neuroprotective Properties of PrP
Many studies have claimed that protection against neuronal damage is one of PrPC's raison d'être. Neuroprotection (defined generically as protecting neurons from dysfunction or death) may represent one of the best-supported functions of PrPC. This protection applies to both physiological challenges and to a peculiar yet fascinating paradigm whereby the closest homolog of PrPC, Dpl, when ectopically expressed in the brain causes loss of Purkinje neurons in the cerebellum but only in a PrP KO background.Citation27
We will begin our discussion of PrP's protective properties with one of the most agreed upon observations-PrP KOs are much more susceptible to ischemic damage. McLennan and colleagues were the first to document that PrP KOs are more susceptible to stroke.Citation81 They were led to the PrP KO through studying a dramatic upregulation of PrP expression at sites of stroke in human brains.Citation81 Subsequent studies have replicated and extended these results in acuteCitation82 and long term models of ischemiaCitation83 and even in transgenic rats.Citation84 Interestingly, transgenic overexpression of PrP in the mouse does not protect above wild-type PrP levelsCitation85 while in the rat increasing PrP levels did confer protection. Weise and colleagues note that PrP KOs have lower levels of phosphorylated-Akt both in basal conditions and during ischemic injury, pointing towards a general role of PrP in activation of cell survival pathways.Citation83 Other researchers have noted significant increases in the phosphorylation of ERK-1 and -2, STAT-1, and JNK-1 in ischemic PrP KO brains.Citation85 Recently, Gains and colleagues have extended PrP's neuroprotective spectrum. They dosed neonatal PrP KOs and controls with a high dose of ethanol, a paradigm for inducing Bax mediated apoptosis, and noted a dramatic increase in cell death in brains of PrP KOs.Citation86 Another brief report documents an enhanced brain injury in PrP KOs after head trauma,Citation87 however, it is likely that these mice overexpress Dpl and therefore display a confounding effect. To test PrP's neuroprotective function in another setting, we crossed PrP KOs to several transgenic models of neurodegenerative disease—Huntington's, Parkinson's and Alzheimer's disease. Much to our surprise, the phenotypes of these diseases were largely unaltered by PrP deletion (ADS, Z. Zhou, W. Jackson, M. Moskowitz, S. Lindquist, unpublished). Thus, the wide-ranging neuroprotective functions of PrP have limitations and these observations of protection in unique models need to be understood in mechanistic detail.
The second well-studied paradigm in which PrP exerts a protective function deals with the neurotoxicity induced by its nearest homolog. The exciting and circuitous discovery of Dpl began with conflicting reports on the phenotype of the PrP KO, with two groups reporting no phenotypeCitation10,Citation22 and one group reporting a late onset ataxia and Purkinje cell loss in Nagasaki PrP KO mice.Citation27,Citation88 Later it was determined by several groups that a previously undescribed tightly linked homolog of PrPC (called “doppel” for “Downstream of the Prnp locus”) was upregulated by the deletion strategy used in the Nagasaki PrP KO line. The Nagasaki deletion strategy fused PrP's promoter to Dpl, driving expression of Dpl in the brain, where it is not normally expressed.Citation89,Citation90 Subsequent experiments determined that the toxicity induced by ectopic Dpl expression in the PrP KO is abrogated by reintroduction of a single copy of PrPC.Citation91,Citation92 However, both Weissmann and Katamine have shown that PrPC cannot suppress higher amounts of transgene driven Dpl expression,Citation93,Citation94 suggesting that Dpl and PrPC are competitive antagonists and can be stoichiometrically titrated against each other. Interestingly, PrPC devoid of the octapeptide repeats (amino acids 23–88) is incapable of rescuing Dpl toxicity, suggesting an important role for the N-proximal region of PrPC in its neuroprotective functions.Citation95 However, a trafficking defect in this deletion mutant of PrP was not ruled out, so PrP may not be reaching the cell surface in these transgenic mice.
A similar phenomenon is observed when an artificial deletion mutant of PrPC, missing amino acids 32–134 and termed “ΔF”, is expressed as a transgene in the PrP KO. This trangenic mouse develops a dramatic loss of granular neurons in the cerebellum, referred to as “Shmerling syndrome” ().Citation96 The deletions of amino acids 32–121 or 32–134, but not shorter deletions of the N-terminus, caused this phenotype. The “ΔF” truncated PrPC targeted specifically to Purkinje cells causes these cells to die in a PrP KO background showing that this is a cell-autonomous phenomenon akin to Dpl toxicity.Citation97 It has also become clear that truncated PrPC and Dpl are toxic to myelinated cells in a PrP KO background.Citation98 The white matter pathology was detected in the cerebellum, brainstem, and spinal cord in “ΔF” mice and extended more broadly in Dpl overexpression where it was also found in the forebrain, pyramidal projections, and the corpus callosum.Citation98 Further deletion mapping of PrPC has narrowed the critical region to forty amino acids in the middle of PrP, 94–134. When PrP lacking these amino acids is expressed in PrP KOs it results in a severe motor phenotype brought about by extensive central and peripheral myelin degeneration ().Citation99 PrPC lacking the octapeptide repeats was able to rescue this deletion mutant induced phenotype unlike the case of Dpl toxicity discussed above.
Most interestingly, careful histopathological analysis clearly identified a myelin degeneration phenotype in PrP KO mice.Citation91,Citation99 Since this phenotype is seen both in Zurich I and in Nagasaki mice, it cannot result from spurious overexpression of Dpl and may indeed represent a consequence of PrP deficiency. The similarity of this phenotype to Shmerling's syndrome is striking—although Shmerling's syndrome is much more severe and is visible in much younger mice. This finding suggests that myelin maintenance may represent an important physiological function of PrPC, and that the defects seen in Shmerling's disease may represent an exaggerated form of a PrPC deficiency syndrome.
In the studies discussed above, all toxic mutants displayed disruption of the charge cluster (CC, residues 95–110) and a part of the hydrophobic core (HC, residues 111–121) of PrP (). Toxicity was ameliorated by co-expressing PrP variants with intact CC and HC, even if these variants lacked the octarepeat region. We therefore posit that PrP exerts its neuroprotective activity by signalling through the central domain to an unknown receptor (tentatively termed PrPR). In all paradigms investigated, the phenotype was determined by the stoichiometry of mutant to full-length PrP, suggesting that PrP, any of the various PrP mutants, and PrPR form hetero-oligomeric complexes (). The mild pathological phenotype of PrP KO mice suggests that myelin integrity is supported by residual PrPR activity, whereas disruption of the central domain (CD) domain sequesters PrPR in a dominant-negative state. Complex stability could be influenced by domains distinct from those involved in executing signal transduction: the context-dependent toxicity of PrP missing part of the hydrophobic core (PrPΔpHC) implicates the octapeptide repreat as one such domain.Citation99 Although deletion of 40 amino acids produced a powerfully toxic molecule, ablation of eight amino acids within this domain (PrPΔpHC) was innocuous to both wild-type and PrP KO mice. Crossing experiments show that PrPΔpHC is not functionally equivalent to PrP. The toxicity of PrPΔF was diminished, yet that of PrPΔCD was augmented by coexpression of PrPΔpHC. In the frame of the signaling model, the deletion in PrPΔpHC may affect the interaction between PrP and PrPR. Verification of the model presented above requires the physical identification of PrPR. Towards that goal, it will be crucial to identify the cellular constituents, which may not necessarily all consist of protein, binding differentially to PrPC.
A Role for PrP in Stem/Progenitor Cell Biology
Recently, it was demonstrated that PrPC is expressed on the surface of hematopoietic stem cells.Citation100 Zhang and colleagues then challenged PrP KO bone marrow with serial transplantations into lethally irradiated recipient mice. After several transplantations, the repopulating potential of PrP KO bone marrow was exhausted whereas control bone marrow was still competent to repopulate lethally irradiated recipient mice, demonstrating that PrP KO hematopoietic stem cells were deficient in “self-renewal”.Citation100 Reintroduction of PrP into PrP null bone marrow cells rescued this defect in self-renewal, arguing against an artifact of genetic background. The molecular pathway by which PrP promotes the self-renewal of hematopoietic stem cells remains unclear but is consistent with many of the suggested protective functions of PrPC. The studies of PrPC in hematopoietic stem cells prompted an examination of PrPC was expression/function in neural stem/precursor cells in the adult brain. Adult neurogenesis is normally neuroanatomically restricted to the dentate gyrus of the hippocampus and the subventricular zone.Citation101 We pulse labeled BrdU into PrP KO and overexpression transgenics and noted that in the dentate gyrus PrP KOs had a lower level of proliferating cells whereas in the PrPC overexpression transgenic cell proliferation was enhanced in the subventricular zone.Citation102 Culturing of neural progenitor cells from embryonic PrP KOs and overexpression transgenics revealed that PrPC may promote the exit from a precursor state and maturation into a neuronal lineage.Citation102 The signaling pathway through which PrPC presumably operates in this process is still undetermined. Collectively, these results demonstrate that careful scrutiny reveals a subtle function for PrPC in stem and precursor cell biology and it will be interesting to examine whether PrPC functions in other adult stem cell populations that can be more readily isolated. In concert with the possible protection against seizure, ischemia, pathogen infection, and evolutionary conservation of PrPC, these results add to the evidence for its relevance to mammalian physiology.
Future Prospects for Determining PrP Function from in Vivo Studies: Beyond the Prion Protein Knockout Mouse
Several investigators have expressed PrPC in yeast in order to better understand the disease-associated properties of this protein.Citation103 Furthermore PrPC has been expressed in yeast (which do not express a PrP homolog) to investigate normal function, but perhaps this is aiming too “low” in terms of model organisms as initial studies have not revealed any role for PrPC in copper transport, a well characterized process in yeast.Citation104 Interestingly PrPC expression rescues Bax induced cell death in yeast;Citation105 however the significance and relevance of this paradigm is unclear even in mammalian cells.Citation106 Given the remarkable conservation of PrP structure among vertebrates,Citation107,Citation108 the use of non-mammalian models could open up new avenues for prion research. For instance, the first dramatic phenotypes of PrP loss- and gain-of-function have been produced in zebrafish (Edward Málaga-Trillo, Gonzalo Solis, Yvonne Schrock, Lydia Luncz, Venus Thomanetz, and Claudia Stuermer, personal communication). Notably, early knockdown of PrP in fish embryos is lethal but can be partially rescued by expression of mouse PrP, ruling out potential off target effects of the knockdown construct. Analysis of the molecular pathways involved in fish PrP function should guide future studies in the mammalian system. Studies in the rat offer the advantage of larger brains that make surgical and other interventions more feasible.Citation77 However, it is unlikely that deletion of PrPC will be much more informative in the rat than it has been for the mouse. Very recently it has become possible to study PrPC function in ruminants lacking PrPC which appear normal up to 20 months of age.Citation11 Also, hemizygous PrPC deletion goats now exist and homozygous null animals should be forthcoming.Citation12 Perhaps the longer lifespans of the cow and goat will reveal age dependent phenotypes associated with PrPC deletion. Likewise it may be worthwhile to delete Prnp in a non-human primate, where much more detailed cognitive testing could be conducted. That being said, it remains possible that the PrP KO mouse holds the key to understanding PrPC function, perhaps even through the study of unexpected phenotypes, such as the recently reported tooth abnormality in PrP KOs.Citation109
Looking to PrPC's closest homologue in the mouse, Dpl, may be fruitful for determining the function of PrPC. Interestingly, the Dpl KO results in male sterility,Citation110,Citation111 a phenotype that can be rescued by testis specific expression of a PrP with an N-terminal deletion in the testes suggesting a functional equivalence of Dpl and the C terminus of PrP (A. Aguzzi, unpublished results). Deletion of both PrPC and Dpl (dKO) had no discernable phenotypes aside from the male infertility present in Dpl KOs.Citation112,Citation113 Deciphering the pathway through which Dpl affects male fertility could inform future studies of PrPC function. A more distant cousin of PrP, called “Shadoo”Citation114 may also hold promise for revealing PrPC's function.
Will the transgenic mice expressing PrPC without a GPI anchor (GPI-) prove useful for elucidating the normal or the disease function of PrP? These mice are very intriguing because replication of prion infectivity is more or less unaffected whereas toxicity is significantly suppressed in the PrP KO background.Citation115,Citation116 It may be that the hypothesized signal transduction properties of PrPC are defective without a GPI anchor attachment, leaving the GPI—mice fully competent for PrPC's only ironclad function—prion replication. This mouse provides an excellent tool in distinguishing PrPSc replication from toxicity.
There are many unresolved questions with respect to PrPC function: What is the connection between PrP function and pathogenesis? What is the functional relevance of differential expression of PrPC in subpopulations of neurons in the brain? How do we assign the many described phenotypes in PrP KOs to discrete molecular pathways? Perhaps physiological stress will only bring out the effects of non-relevant linked genes, leading down a garden path of genetic artifacts? Is the conformational diversity attained by PrPSc reflecting a possible conformational diversity of PrPC as recently speculated by Stanley Prusiner?Citation117 It is fascinating to speculate that this labile structure of PrPC may encipher its function. Hopefully one day the question of PrPC function will be settled, but for now there remains plenty of work to do!
Abbreviations
| PrP | = | prion protein |
| KO | = | knockout |
| GPI | = | glycosylphosphatidylinositol |
| Cu | = | copper |
| CC | = | charge cluster |
| HC | = | hydrophobic core |
| SOD | = | superoxide dismutase |
Figures and Tables
Figure 1 The overlap between the normal function of PrPC and the pathogenic dysfunction of PrPSc in disease depicted as a Venn diagram. The extent of overlap between the normal function of PrPC and its role in prion disease is open to speculation, the circles could have a much greater or perhaps even less overlap.
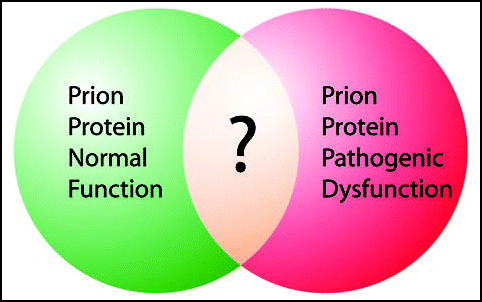
Figure 2 Different approaches to study PrPC's normal function. There are many ways to approach the study of the normal function of PrPC, none of which have conclusively demonstrated PrPC's function. Interacting partners of PrPC have yielded many interesting candidates, human genetic studies have found associations of PrPC with diseases beyond prion disease and even to learning and memory, over- and ectopic-expression studies constitute another approach to determine the function of PrPC, and finally, the focus of this review, the PrP KO has given some clues to the function of PrPC.
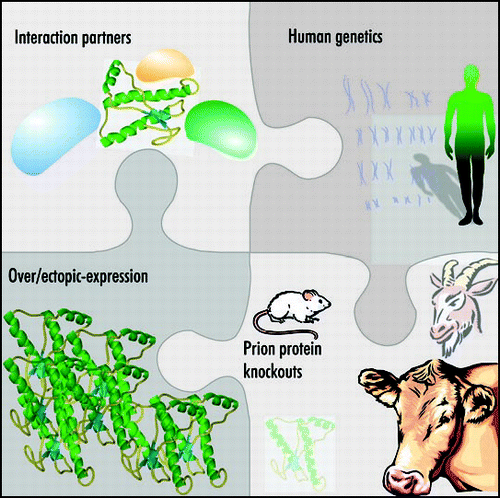
Figure 3 A model for the effects of PrPC deletion and deletion mutants of PrPC. (A) Schematic diagram of wild-type PrPC and deletion mutants. SP, signal peptide; octapeptide repeats are indicated in blue; CC, charge cluster; HC, hydrophobic core; H1, H2, H3 Helix 1,2 and 3, respectively; GFP, GPI-anchor addition sequence (B). PrP (black) consists of a globular C-terminal domain (hexagon) and a N-terminal flexible tail (arch) encompassing the octapeptide repeats (ORs) (circle). The model rests on the following assumptions: (1) PrP activates a hitherto unidentified receptor (PrPR) which transmits myelin maintenance signals (flashes); (2) in the absence of PrP, PrPR exerts some residual activity, either constitutively or by recruiting a surrogate ligand; (3) the activity of PrP and its mutants requires homo- or heterodimerization, and induces dimerization of PrPR; and (4) PrP dimers containing PrPΔCD or PrPΔCD trap PrPR in an inactive dominant-negative state. Finally, (5) the OR region stabilizes the interaction between PrP and PrPR, but does not contribute directly to signaling.
Table 1 Proposed functions for PrP from analysis of PrP knockout mice
Acknowledgements
We are grateful to numerous enthusiastic and collegial PrP function collaborators: Artur Topolszki, Cheng Cheng Zhang, Harvey Lodish, Oliver King, Walker Jackson and Rob Wheeler (WIBR), Jason Emsley, Hande Ozdinler, Jeffrey Macklis, Zhipeng Zhou, Katharina Haerter, Michael Moskowitz and Carla Bender Kim (Harvard Medical School), to Tom DiCesare of WIBR bioinformatics for graphical assistance, to Caroline Yi (Harvard Medical School), Walker Jackson (WIBR) and Edward Málaga-Trillo (University of Konstanz) for valuable comments on the manuscript. Susan Lindquist is a Howard Hughes Medical Institute Investigator and is supported by funding from the Ellison Medical Research Foundation, U.S. Department of Defense and the NIH. Adriano Aguzzi is supported by a philanthropic donation by Dr. Arthur Meier-Schenk, and by grants from the European Union (TSEUR), the Swiss National Foundation, the Ernst-Jung Foundation and the National Competence Center for Research on Neural Plasticity and Repair. We apologize to authors whose work could not be discussed due to limitations in space and scope.
References
- Prusiner SB. Prions. Proc Natl Acad Sci USA 1998; 95:13363 - 13383
- Aguzzi A, Heikenwalder M. Pathogenesis of prion diseases: Current status and future outlook. Nat Rev Microbiol 2006; 4:765 - 775
- Hetz C, Maundrell K, Soto C. Is loss of function of the prion protein the cause of prion disorders?. Trends Mol Med 2003; 9:237 - 243
- Aguzzi A. Prion diseases of humans and farm animals: Epidemiology, genetics, and pathogenesis. J Neurochem 2006; 97:1726 - 1739
- Caughey B, Baron GS. Prions and their partners in crime. Nature 2006; 443:803 - 810
- Sakudo A, Onodera T, Suganuma Y, Kobayashi T, Saeki K, Ikuta K. Recent advances in clarifying prion protein functions using knockout mice and derived cell lines. Mini Rev Med Chem 2006; 6:589 - 601
- Aguzzi A, Haass C. Games played by rogue proteins in prion disorders and Alzheimer's disease. Science 2003; 302:814 - 818
- Abeliovich A, Schmitz Y, Farinas I, Choi-Lundberg D, Ho WH, Castillo PE, Shinsky N, Verdugo JM, Armanini M, Ryan A, Hynes M, Phillips H, Sulzer D, Rosenthal A. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 2000; 25:239 - 252
- Zuccato C, Ciammola A, Rigamonti D, Leavitt BR, Goffredo D, Conti L, MacDonald ME, Friedlander RM, Silani V, Hayden MR, Timmusk T, Sipione S, Cattaneo E. Loss of huntingtin-mediated BDNF gene transcription in Huntington's disease. Science 2001; 293:493 - 498
- Bueler H, Fischer M, Lang Y, Bluethmann H, Lipp HP, DeArmond SJ, Prusiner SB, Aguet M, Weissmann C. Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature 1992; 356:577 - 582
- Richt JA, Kasinathan P, Hamir AN, Castilla J, Sathiyaseelan T, Vargas F, Sathiyaseelan J, Wu H, Matsushita H, Koster J, Kato S, Ishida I, Soto C, Robl JM, Kuroiwa Y. Production of cattle lacking prion protein. Nat Biotechnol 2006;
- Yu G, Chen J, Yu H, Liu S, Xu X, Sha H, Zhang X, Wu G, Xu S, Cheng G. Functional disruption of the prion protein gene in cloned goats. J Gen Virol 2006; 87:1019 - 1027
- Marc D, Mercey R, Lantier F. Scavenger, transducer, RNA chaperone? What ligands of the prion protein teach us about its function. Cell Mol Life Sci 2007;
- Riemenschneider M, Klopp N, Xiang W, Wagenpfeil S, Vollmert C, Muller U, Forstl H, Illig T, Kretzschmar H, Kurz A. Prion protein codon 129 polymorphism and risk of Alzheimer disease. Neurology 2004; 63:364 - 366
- Ahn K, Kim E, Kwon YA, Kim DK, Lee JE, Jo SA. No association of prion protein gene polymorphisms with Alzheimer' s disease in Korean population. Exp Mol Med 2006; 38:727 - 731
- Del Bo R, Scarlato M, Ghezzi S, Martinelli-Boneschi F, Fenoglio C, Galimberti G, Galbiati S, Virgilio R, Galimberti D, Ferrarese C, Scarpini E, Bresolin N, Comi GP. Is M129V of PRNP gene associated with Alzheimer's disease? A case-control study and a meta-analysis. Neurobiol Aging 2006; 27:770 e1 - 770 e5
- Walz R, Castro RM, Landemberger MC, Velasco TR, Terra-Bustamante VC, Bastos AC, Bianchin M, Wichert-Ana L, Araujo D, Alexandre V Jr, Santos AC, Machado HR, Carlotti CG Jr, Brentani RR, Martins VR, Sakamoto AC. Cortical malformations are associated with a rare polymorphism of cellular prion protein. Neurology 2004; 63:557 - 560
- Walz R, Castro RM, Velasco TR, Alexandre V Jr, Lopes MH, Leite JP, Santos AC, Assirati JA Jr, Wichert-Ana L, Terra-Bustamante VC, Bianchin MM, Maciag PC, Ribeiro KB, Guarnieri R, Araujo D, Cabalero O, Moura R, Salim AC, Kindlmann K, Landemberger MC, Marques W Jr, Fernandes RM, Serafini LN, Machado HR, Carlotti CG Jr, Brentani RR, Sakamoto AC, Martins VR. Surgical outcome in mesial temporal sclerosis correlates with prion protein gene variant. Neurology 2003; 61:1204 - 1210
- Papassotiropoulos A, Wollmer MA, Aguzzi A, Hock C, Nitsch RM, de Quervain DJ. The prion gene is associated with human long-term memory. Hum Mol Genet 2005; 14:2241 - 2246
- Zan Q, Wen B, He Y, Wang Y, Xu S, Qian J, Lu D, Jin L. Complete sequence data support lack of balancing selection on PRNP in a natural Chinese population. J Hum Genet 2006; 51:451 - 454
- Weissmann C, Bueler H. A mouse to remember. Cell 2004; 116:S111 - S113
- Manson JC, Clarke AR, Hooper ML, Aitchison L, McConnell I, Hope J. 129/Ola mice carrying a null mutation in PrP that abolishes mRNA production are developmentally normal. Mol Neurobiol 1994; 8:121 - 127
- Roucou X, Gains M, LeBlanc AC. Neuroprotective functions of prion protein. J Neurosci Res 2004; 75:153 - 161
- Weissmann C, Flechsig E. PrP knock-out and PrP transgenic mice in prion research. Br Med Bull 2003; 66:43 - 60
- Weissmann C, Aguzzi A. Perspectives: Neurobiology: PrP's double causes trouble. Science 1999; 286:914 - 915
- Gerlai R. Gene-targeting studies of mammalian behavior: Is it the mutation or the background genotype?. Trends Neurosci 1996; 19:177 - 181
- Sakaguchi S, Katamine S, Nishida N, Moriuchi R, Shigematsu K, Sugimoto T, Nakatani A, Kataoka Y, Houtani T, Shirabe S, Okada H, Hasegawa S, Miyamoto T, Noda T. Loss of cerebellar Purkinje cells in aged mice homozygous for a disrupted PrP gene. Nature 1996; 380:528 - 531
- Bueler H, Aguzzi A, Sailer A, Greiner RA, Autenried P, Aguet M, Weissmann C. Mice devoid of PrP are resistant to scrapie. Cell 1993; 73:1339 - 1347
- Sailer A, Bueler H, Fischer M, Aguzzi A, Weissmann C. No propagation of prions in mice devoid of PrP. Cell 1994; 77:967 - 968
- Bueler H, Raeber A, Sailer A, Fischer M, Aguzzi A, Weissmann C. High prion and PrPSc levels but delayed onset of disease in scrapie-inoculated mice heterozygous for a disrupted PrP gene. Mol Med 1994; 1:19 - 30
- Brandner S, Isenmann S, Raeber A, Fischer M, Sailer A, Kobayashi Y, Marino S, Weissmann C, Aguzzi A. Normal host prion protein necessary for scrapie-induced neurotoxicity. Nature 1996; 379:339 - 343
- Fischer M, Rulicke T, Raeber A, Sailer A, Moser M, Oesch B, Brandner S, Aguzzi A, Weissmann C. Prion protein (PrP) with amino-proximal deletions restoring susceptibility of PrP knockout mice to scrapie. Embo J 1996; 15:1255 - 1264
- Flechsig E, Shmerling D, Hegyi I, Raeber AJ, Fischer M, Cozzio A, von Mering C, Aguzzi A, Weissmann C. Prion protein devoid of the octapeptide repeat region restores susceptibility to scrapie in PrP knockout mice. Neuron 2000; 27:399 - 408
- Tobler I, Gaus SE, Deboer T, Achermann P, Fischer M, Rulicke T, Moser M, Oesch B, McBride PA, Manson JC. Altered circadian activity rhythms and sleep in mice devoid of prion protein. Nature 1996; 380:639 - 642
- Lugaresi E, Tobler I, Gambetti P, Montagna P. The pathophysiology of fatal familial insomnia. Brain Pathol 1998; 8:521 - 526
- Landolt HP, Glatzel M, Blattler T, Achermann P, Roth C, Mathis J, Weis J, Tobler I, Aguzzi A, Bassetti CL. Sleep-wake disturbances in sporadic Creutzfeldt-Jakob disease. Neurology 2006; 66:1418 - 1424
- Tobler I, Deboer T, Fischer M. Sleep and sleep regulation in normal and prion protein-deficient mice. J Neurosci 1997; 17:1869 - 1879
- Brown DR, Qin K, Herms JW, Madlung A, Manson J, Strome R, Fraser PE, Kruck T, von Bohlen A, Schulz-Schaeffer W, Giese A, Westaway D, Kretzschmar H. The cellular prion protein binds copper in vivo. Nature 1997; 390:684 - 687
- Waggoner DJ, Drisaldi B, Bartnikas TB, Casareno RL, Prohaska JR, Gitlin JD, Harris DA. Brain copper content and cuproenzyme activity do not vary with prion protein expression level. J Biol Chem 2000; 275:7455 - 7458
- Wong BS, Liu T, Li R, Pan T, Petersen RB, Smith MA, Gambetti P, Perry G, Manson JC, Brown DR, Sy MS. Increased levels of oxidative stress markers detected in the brains of mice devoid of prion protein. J Neurochem 2001; 76:565 - 572
- Klamt F, Dal-Pizzol F, Conte da Frota MJ, Walz R, Andrades ME, da Silva EG, Brentani RR, Izquierdo I, Fonseca Moreira JC. Imbalance of antioxidant defense in mice lacking cellular prion protein. Free Radic Biol Med 2001; 30:1137 - 1144
- Brown DR, Nicholas RS, Canevari L. Lack of prion protein expression results in a neuronal phenotype sensitive to stress. J Neurosci Res 2002; 67:211 - 224
- Hutter G, Heppner FL, Aguzzi A. No superoxide dismutase activity of cellular prion protein in vivo. Biol Chem 2003; 384:1279 - 1285
- Miele G, Jeffrey M, Turnbull D, Manson J, Clinton M. Ablation of cellular prion protein expression affects mitochondrial numbers and morphology. Biochem Biophys Res Commun 2002; 291:372 - 377
- Ma J, Wollmann R, Lindquist S. Neurotoxicity and neurodegeneration when PrP accumulates in the cytosol. Science 2002; 298:1781 - 1785
- Lobao-Soares B, Bianchin MM, Linhares MN, Carqueja CL, Tasca CI, Souza M, Marques W Jr, Brentani R, Martins VR, Sakamoto AC, Carlotti CG Jr, Walz R. Normal brain mitochondrial respiration in adult mice lacking cellular prion protein. Neurosci Lett 2005; 375:203 - 206
- de Almeida CJ, Chiarini LB, da Silva JP, PM ES, Martins MA, Linden R. The cellular prion protein modulates phagocytosis and inflammatory response. J Leukoc Biol 2005; 77:238 - 246
- Watarai M, Kim S, Erdenebaatar J, Makino S, Horiuchi M, Shirahata T, Sakaguchi S, Katamine S. Cellular prion protein promotes Brucella infection into macrophages. J Exp Med 2003; 198:5 - 17
- Aguzzi A, Hardt WD. Dangerous liaisons between a microbe and the prion protein. J Exp Med 2003; 198:1 - 4
- Fontes P, Alvarez-Martinez MT, Gross A, Carnaud C, Kohler S, Liautard JP. Absence of evidence for the participation of the macrophage cellular prion protein in infection with Brucella suis. Infect Immun 2005; 73:6229 - 6236
- Thackray AM, Bujdoso R. PrPC expression influences the establishment of herpes simplex virus type 1 latency. J Virol 2002; 76:2498 - 2509
- Thackray AM, Bujdoso R. Elevated PrPC expression predisposes to increased HSV-1 pathogenicity. Antivir Chem Chemother 2006; 17:41 - 52
- Ballerini C, Gourdain P, Bachy V, Blanchard N, Levavasseur E, Gregoire S, Fontes P, Aucouturier P, Hivroz C, Carnaud C. Functional implication of cellular prion protein in antigen-driven interactions between T cells and dendritic cells. J Immunol 2006; 176:7254 - 7262
- Jouvin-Marche E, Attuil-Audenis V, Aude-Garcia C, Rachidi W, Zabel M, Podevin-Dimster V, Siret C, Huber C, Martinic M, Riondel J, Villiers CL, Favier A, Naquet P, Cesbron JY, Marche PN. Overexpression of cellular prion protein induces an antioxidant environment altering T cell development in the thymus. J Immunol 2006; 176:3490 - 3497
- Kubosaki A, Nishimura-Nasu Y, Nishimura T, Yusa S, Sakudo A, Saeki K, Matsumoto Y, Itohara S, Onodera T. Expression of normal cellular prion protein (PrPC) on T lymphocytes and the effect of copper ion: Analysis by wild-type and prion protein gene-deficient mice. Biochem Biophys Res Commun 2003; 307:810 - 813
- Collinge J, Whittington MA, Sidle KC, Smith CJ, Palmer MS, Clarke AR, Jefferys JG. Prion protein is necessary for normal synaptic function. Nature 1994; 370:295 - 297
- Manson JC CA, Johnston A, Black C, MacLeod N. PrP Gene dosage and long term potentiation. Neurodegeneration 1995; 4:113 - 114
- Whittington MA, Sidle KC, Gowland I, Meads J, Hill AF, Palmer MS, Jefferys JG, Collinge J. Rescue of neurophysiological phenotype seen in PrP null mice by transgene encoding human prion protein. Nat Genet 1995; 9:197 - 201
- Asante EA, Li YG, Gowland I, Jefferys JG, Collinge J. Pathogenic human prion protein rescues PrP null phenotype in transgenic mice. Neurosci Lett 2004; 360:33 - 36
- Herms JW, Kretzchmar HA, Titz S, Keller BU. Patch-clamp analysis of synaptic transmission to cerebellar purkinje cells of prion protein knockout mice. Eur J Neurosci 1995; 7:2508 - 2512
- Lledo PM, Tremblay P, DeArmond SJ, Prusiner SB, Nicoll RA. Mice deficient for prion protein exhibit normal neuronal excitability and synaptic transmission in the hippocampus. Proc Natl Acad Sci USA 1996; 93:2403 - 2407
- Criado JR, Sanchez-Alavez M, Conti B, Giacchino JL, Wills DN, Henriksen SJ, Race R, Manson JC, Chesebro B, Oldstone MB. Mice devoid of prion protein have cognitive deficits that are rescued by reconstitution of PrP in neurons. Neurobiol Dis 2005; 19:255 - 265
- Maglio LE, Perez MF, Martins VR, Brentani RR, Ramirez OA. Hippocampal synaptic plasticity in mice devoid of cellular prion protein. Brain Res Mol Brain Res 2004; 131:58 - 64
- Curtis J, Errington M, Bliss T, Voss K, MacLeod N. Age-dependent loss of PTP and LTP in the hippocampus of PrP-null mice. Neurobiol Dis 2003; 13:55 - 62
- Maglio LE, Martins VR, Izquierdo I, Ramirez OA. Role of cellular prion protein on LTP expression in aged mice. Brain Res 2006; 1097:11 - 18
- Herms JW, Tings T, Dunker S, Kretzschmar HA. Prion protein affects Ca2+-activated K+ currents in cerebellar purkinje cells. Neurobiol Dis 2001; 8:324 - 330
- Carleton A, Tremblay P, Vincent JD, Lledo PM. Dose-dependent, prion protein (PrP)-mediated facilitation of excitatory synaptic transmission in the mouse hippocampus. Pflugers Arch 2001; 442:223 - 229
- Mallucci GR, Ratte S, Asante EA, Linehan J, Gowland I, Jefferys JG, Collinge J. Post-natal knockout of prion protein alters hippocampal CA1 properties, but does not result in neurodegeneration. Embo J 2002; 21:202 - 210
- Walz R, Amaral OB, Rockenbach IC, Roesler R, Izquierdo I, Cavalheiro EA, Martins VR, Brentani RR. Increased sensitivity to seizures in mice lacking cellular prion protein. Epilepsia 1999; 40:1679 - 1682
- Rangel A, Burgaya F, Gavin R, Soriano E, Aguzzi A, Del Rio JA. Enhanced susceptibility of Prnp-deficient mice to kainate-induced seizures, neuronal apoptosis, and death: Role of AMPA/kainate receptors. J Neurosci Res 2007;
- Pereira GS, Walz R, Bonan CD, Battastini AM, Izquierdo I, Martins VR, Brentani RR, Sarkis JJ. Changes in cortical and hippocampal ectonucleotidase activities in mice lacking cellular prion protein. Neurosci Lett 2001; 301:72 - 74
- Walz R, Castro RM, Velasco TR, Carlotti CG Jr, Sakamoto AC, Brentani RR, Martins VR. Cellular prion protein: Implications in seizures and epilepsy. Cell Mol Neurobiol 2002; 22:249 - 257
- Colling SB, Khana M, Collinge J, Jefferys JG. Mossy fibre reorganization in the hippocampus of prion protein null mice. Brain Res 1997; 755:28 - 35
- Lipp HP, Stagliar-Bozicevic M, Fischer M, Wolfer DP. A 2-year longitudinal study of swimming navigation in mice devoid of the prion protein: No evidence for neurological anomalies or spatial learning impairments. Behav Brain Res 1998; 95:47 - 54
- Roesler R, Walz R, Quevedo J, de-Paris F, Zanata SM, Graner E, Izquierdo I, Martins VR, Brentani RR. Normal inhibitory avoidance learning and anxiety, but increased locomotor activity in mice devoid of PrPC. Brain Res Mol Brain Res 1999; 71:349 - 353
- Coitinho AS, Roesler R, Martins VR, Brentani RR, Izquierdo I. Cellular prion protein ablation impairs behavior as a function of age. Neuroreport 2003; 14:1375 - 1379
- Coitinho AS, Freitas AR, Lopes MH, Hajj GN, Roesler R, Walz R, Rossato JI, Cammarota M, Izquierdo I, Martins VR, Brentani RR. The interaction between prion protein and laminin modulates memory consolidation. Eur J Neurosci 2006; 24:3255 - 3264
- Coitinho AS, Dietrich MO, Hoffmann A, Dall'Igna OP, Souza DO, Martins VR, Brentani RR, Izquierdo I, Lara DR. Decreased hyperlocomotion induced by MK-801, but not amphetamine and caffeine in mice lacking cellular prion protein (PrPC). Brain Res Mol Brain Res 2002; 107:190 - 194
- Nico PB, de-Paris F, Vinade ER, Amaral OB, Rockenbach I, Soares BL, Guarnieri R, Wichert-Ana L, Calvo F, Walz R, Izquierdo I, Sakamoto AC, Brentani R, Martins VR, Bianchin MM. Altered behavioural response to acute stress in mice lacking cellular prion protein. Behav Brain Res 2005; 162:173 - 181
- Steele AD, Jackson WS, King OD, Lindquist S. The power of automated high-resolution behavior analysis revealed by its application to mouse models of Huntington's and prion diseases. Proc Natl Acad Sci USA 2007; 104:1983 - 1988
- McLennan NF, Brennan PM, McNeill A, Davies I, Fotheringham A, Rennison KA, Ritchie D, Brannan F, Head MW, Ironside JW, Williams A, Bell JE. Prion protein accumulation and neuroprotection in hypoxic brain damage. Am J Pathol 2004; 165:227 - 235
- Sakurai-Yamashita Y, Sakaguchi S, Yoshikawa D, Okimura N, Masuda Y, Katamine S, Niwa M. Female-specific neuroprotection against transient brain ischemia observed in mice devoid of prion protein is abolished by ectopic expression of prion protein-like protein. Neuroscience 2005; 136:281 - 287
- Weise J, Sandau R, Schwarting S, Crome O, Wrede A, Schulz-Schaeffer W, Zerr I, Bahr M. Deletion of cellular prion protein results in reduced Akt activation, enhanced postischemic caspase-3 activation, and exacerbation of ischemic brain injury. Stroke 2006; 37:1296 - 1300
- Shyu WC, Lin SZ, Chiang MF, Ding DC, Li KW, Chen SF, Yang HI, Li H. Overexpression of PrPC by adenovirus-mediated gene targeting reduces ischemic injury in a stroke rat model. J Neurosci 2005; 25:8967 - 8977
- Spudich A, Frigg R, Kilic E, Kilic U, Oesch B, Raeber A, Bassetti CL, Hermann DM. Aggravation of ischemic brain injury by prion protein deficiency: Role of ERK-1/-2 and STAT-1. Neurobiol Dis 2005; 20:442 - 449
- Gains MJ, Roth KA, LeBlanc AC. Prion protein protects against ethanol-induced Bax-mediated cell death in vivo. Neuroreport 2006; 17:903 - 906
- Hoshino S, Inoue K, Yokoyama T, Kobayashi S, Asakura T, Teramoto A, Itohara S. Prions prevent brain damage after experimental brain injury: A preliminary report. Acta Neurochir Suppl 2003; 86:297 - 299
- Katamine S, Nishida N, Sugimoto T, Noda T, Sakaguchi S, Shigematsu K, Kataoka Y, Nakatani A, Hasegawa S, Moriuchi R, Miyamoto T. Impaired motor coordination in mice lacking prion protein. Cell Mol Neurobiol 1998; 18:731 - 742
- Moore RC, Lee IY, Silverman GL, Harrison PM, Strome R, Heinrich C, Karunaratne A, Pasternak SH, Chishti MA, Liang Y, Mastrangelo P, Wang K, Smit AF, Katamine S, Carlson GA, Cohen FE, Prusiner SB, Melton DW, Tremblay P, Hood LE, Westaway D. Ataxia in prion protein (PrP)-deficient mice is associated with upregulation of the novel PrP-like protein doppel. J Mol Biol 1999; 292:797 - 817
- Li A, Sakaguchi S, Atarashi R, Roy BC, Nakaoke R, Arima K, Okimura N, Kopacek J, Shigematsu K. Identification of a novel gene encoding a PrP-like protein expressed as chimeric transcripts fused to PrP exon 1/2 in ataxic mouse line with a disrupted PrP gene. Cell Mol Neurobiol 2000; 20:553 - 567
- Nishida N, Tremblay P, Sugimoto T, Shigematsu K, Shirabe S, Petromilli C, Erpel SP, Nakaoke R, Atarashi R, Houtani T, Torchia M, Sakaguchi S, DeArmond SJ, Prusiner SB, Katamine S. A mouse prion protein transgene rescues mice deficient for the prion protein gene from purkinje cell degeneration and demyelination. Lab Invest 1999; 79:689 - 697
- Valenti P, Cozzio A, Nishida N, Wolfer DP, Sakaguchi S, Lipp HP. Similar target, different effects: Late-onset ataxia and spatial learning in prion protein-deficient mouse lines. Neurogenetics 2001; 3:173 - 184
- Anderson L, Rossi D, Linehan J, Brandner S, Weissmann C. Transgene-driven expression of the Doppel protein in Purkinje cells causes Purkinje cell degeneration and motor impairment. Proc Natl Acad Sci USA 2004; 101:3644 - 3649
- Yamaguchi N, Sakaguchi S, Shigematsu K, Okimura N, Katamine S. Doppel-induced Purkinje cell death is stoichiometrically abrogated by prion protein. Biochem Biophys Res Commun 2004; 319:1247 - 1252
- Atarashi R, Nishida N, Shigematsu K, Goto S, Kondo T, Sakaguchi S, Katamine S. Deletion of N-terminal residues 23–88 from prion protein (PrP) abrogates the potential to rescue PrP-deficient mice from PrP-like protein/doppel-induced Neurodegeneration. J Biol Chem 2003; 278:28944 - 28949
- Shmerling D, Hegyi I, Fischer M, Blattler T, Brandner S, Gotz J, Rulicke T, Flechsig E, Cozzio A, von Mering C, Hangartner C, Aguzzi A, Weissmann C. Expression of amino-terminally truncated PrP in the mouse leading to ataxia and specific cerebellar lesions. Cell 1998; 93:203 - 214
- Flechsig E, Hegyi I, Leimeroth R, Zuniga A, Rossi D, Cozzio A, Schwarz P, Rulicke T, Gotz J, Aguzzi A, Weissmann C. Expression of truncated PrP targeted to Purkinje cells of PrP knockout mice causes Purkinje cell death and ataxia. EMBO J 2003; 22:3095 - 3101
- Radovanovic I, Braun N, Giger OT, Mertz K, Miele G, Prinz M, Navarro B, Aguzzi A. Truncated prion protein and Doppel are myelinotoxic in the absence of oligodendrocytic PrPC. J Neurosci 2005; 25:4879 - 4888
- Baumann F, Tolnay M, Brabeck C, Pahnke J, Kloz U, Niemann HH, Heikenwalder M, Rulicke T, Burkle A, Aguzzi A. Lethal recessive myelin toxicity of prion protein lacking its central domain. Embo J 2007; 26:538 - 547
- Zhang CC, Steele AD, Lindquist S, Lodish HF. Prion protein is expressed on long-term repopulating hematopoietic stem cells and is important for their self-renewal. Proc Natl Acad Sci USA 2006; 103:2184 - 2189
- Emsley JG, Mitchell BD, Kempermann G, Macklis JD. Adult neurogenesis and repair of the adult CNS with neural progenitors, precursors, and stem cells. Prog Neurobiol 2005; 75:321 - 341
- Steele AD, Emsley JG, Ozdinler PH, Lindquist S, Macklis JD. Prion protein (PrPC) positively regulates neural precursor proliferation during developmental and adult mammalian neurogenesis. Proc Natl Acad Sci USA 2006; 103:3416 - 3421
- Ma J, Lindquist S. De novo generation of a PrPSc-like conformation in living cells. Nat Cell Biol 1999; 1:358 - 361
- Li A, Dong J, Harris DA. Cell surface expression of the prion protein in yeast does not alter copper utilization phenotypes. J Biol Chem 2004; 279:29469 - 29477
- Li A, Harris DA. Mammalian prion protein suppresses Bax-induced cell death in yeast. J Biol Chem 2005; 280:17430 - 17434
- Bounhar Y, Zhang Y, Goodyer CG, LeBlanc A. Prion protein protects human neurons against Bax-mediated apoptosis. J Biol Chem 2001; 276:39145 - 39149
- Calzolai L, Lysek DA, Perez DR, Guntert P, Wuthrich K. Prion protein NMR structures of chickens, turtles, and frogs. Proc Natl Acad Sci USA 2005; 102:651 - 655
- Rivera-Milla E, Oidtmann B, Panagiotidis CH, Baier M, Sklaviadis T, Hoffmann R, Zhou Y, Solis GP, Stuermer CA, Malaga-Trillo E. Disparate evolution of prion protein domains and the distinct origin of Doppel- and prion-related loci revealed by fish-to-mammal comparisons. Faseb J 2006; 20:317 - 319
- Schneider K, Korkmaz Y, Addicks K, Lang H, Raab WH. Prion Protein (PrP) in human teeth: An unprecedented pointer to PrP's function. J Endod 2007; 33:110 - 113
- Behrens A, Genoud N, Naumann H, Rulicke T, Janett F, Heppner FL, Ledermann B, Aguzzi A. Absence of the prion protein homologue Doppel causes male sterility. EMBO J 2002; 21:3652 - 3658
- Tuzi NL, Gall E, Melton D, Manson JC. Expression of doppel in the CNS of mice does not modulate transmissible spongiform encephalopathy disease. J Gen Virol 2002; 83:705 - 711
- Genoud N, Behrens A, Miele G, Robay D, Heppner FL, Freigang S, Aguzzi A. Disruption of Doppel prevents neurodegeneration in mice with extensive Prnp deletions. Proc Natl Acad Sci USA 2004; 101:4198 - 4203
- Paisley D, Banks S, Selfridge J, McLennan NF, Ritchie AM, McEwan C, Irvine DS, Saunders PT, Manson JC, Melton DW. Male infertility and DNA damage in Doppel knockout and prion protein/Doppel double-knockout mice. Am J Pathol 2004; 164:2279 - 2288
- Premzl M, Sangiorgio L, Strumbo B, Marshall Graves JA, Simonic T, Gready JE. Shadoo, a new protein highly conserved from fish to mammals and with similarity to prion protein. Gene 2003; 314:89 - 102
- Chesebro B, Trifilo M, Race R, Meade-White K, Teng C, LaCasse R, Raymond L, Favara C, Baron G, Priola S, Caughey B, Masliah E, Oldstone M. Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science 2005; 308:1435 - 1439
- Aguzzi A. Cell biology. Prion toxicity: All sail and no anchor. Science 2005; 308:1420 - 1421
- Legname G, Nguyen HO, Peretz D, Cohen FE, DeArmond SJ, Prusiner SB. Continuum of prion protein structures enciphers a multitude of prion isolate-specified phenotypes. Proc Natl Acad Sci USA 2006; 103:19105 - 19110