Abstract
In neurodegenerative disorders of the aging population, misfolded proteins, such as PrPSc, α-synuclein, amyloid β protein and tau, can interact resulting in enhanced aggregation, cross seeding and accelerated disease progression. Previous reports have shown that in Creutzfeldt-Jakob disease and scrapie, α-synuclein accumulates near PrPSc deposits. However, it is unclear if pre-existing human α-synuclein aggregates modified prion disease pathogenesis, or if PrPSc exacerbates the α-synuclein pathology. Here, we inoculated infectious prions into aged α-synuclein transgenic (tg) and non-transgenic littermate control mice by the intracerebral route. Remarkably, inoculation of RML and mNS prions into α-synuclein tg mice resulted in more extensive and abundant intraneuronal and synaptic α-synuclein accumulation. In addition, infectious prions led to the formation of perineuronal α-synuclein deposits with a neuritic plaque-like appearance. Prion pathology was unmodified by the presence of α-synuclein. However, with the mNS prion strain there was a modest but significant acceleration in the time to terminal prion disease in mice having α-synuclein aggregates as compared with non-tg mice. Taken together, these studies support the notion that PrPSc directly or indirectly promotes α-synuclein pathology.
Introduction
Misfolded protein aggregates are pathologic hallmarks for many neurodegenerative disorders, including Alzheimer, Parkinson and prion disease.Citation1 Protein aggregates are typically β-sheet rich oligomers or fibrils that occur as intracellular or extracellular deposits in the central nervous system.Citation2 In patients with neurodegenerative disease, two or more protein aggregates commonly co-exist, for example α-synuclein and amyloid β in dementia with Lewy bodiesCitation3,Citation4 and tau and amyloid β in Alzheimer disease.Citation5 Consistent with these observations, a patient was recently reported that had suffered from a rapidly progressive dementia with multiple aggregated proteins existing in the brain, including prions, tau, α-synuclein and amyloid β,Citation6 which may suggest a general disturbance in protein folding or an initial protein aggregate catalyzing the aggregation of additional proteins.
In vitro, small aggregates or fibrils can “seed” or recruit soluble monomers of the same protein and accelerate fibril formation.Citation7 Cross-seeding experiments further demonstrated that an aggregate nucleus of one protein can induce fibril formation of an unrelated protein.Citation4,Citation8 Whether cross-seeding occurs in vivo is unclear, however the co-occurrence of distinct protein aggregates indicates the possibility that additional proteins are triggered by the initial seed to assemble into fibrils.Citation9,Citation10 A cascade of multiple misfolded proteins may accelerate neurodegeneration and lead to a rapidly progressive clinical disease.Citation11
Infectious prions are the only known protein aggregates that naturally transmit to other individuals.Citation12 In prion disease, the misfolded, aggregated conformer, PrPSc, induces the conversion of the host prion protein, PrPC, in an autocatalytic self-templating process.Citation13 Prion conversion spreads throughout the brain inciting massive astrogliosis, neuronal loss, and spongiform degeneration.Citation14 Once evident, clinical signs are rapidly progressive and ultimately fatal.
With regards to α-synuclein, protein misfolding and aggregation into protofibrilsCitation15 and oligomers has been shown to lead to neurodegeneration.Citation16 In addition, recent evidence suggest that prion-like propagation of α-synuclein from neuron to neuronCitation17 and neuron to gliaCitation18 might also play an important role in the neurodegenerative process.Citation19 Although few studies report the co-occurrence of α-synuclein and prions,Citation20,Citation21 both proteins are present in synapses,Citation22,Citation23 and the synaptic loss due to α-synuclein aggregates may delay or accelerate prion disease. It is unclear if prion aggregates can exacerbate α-synuclein pathology in transgenic models of Parkinson-like disease. In prion disease, the incubation period is highly predictable and with little variability in lab animal models, which is advantageous in studying modifiers of disease. In this context, we investigated whether (1) pre-existing human α-synuclein aggregates modified prion disease pathogenesis, or (2) PrPSc exacerbates α-synuclein pathology. We inoculated infectious prions into aged α-synuclein transgenic (tg) and non-transgenic (non-tg) littermate control mice by the intracerebral route. Our studies showed that inoculation of RML and mNS prions into α-synuclein tg mice resulted in more extensive α-synuclein accumulation with formation of perineuronal α-synuclein deposits with a neuritic plaque-like appearance.
Results
Incubation period for the mNS prion strain is accelerated in α-synuclein transgenic mice
We inoculated mouse-adapted natural sheep scrapie (mNS) prions or uninfected brain homogenate into 10-mo-old α-synuclein tg mice and littermate non-transgenic controls to determine whether either disease is modified in these well-characterized models. Mice inoculated with uninfected brain homogenate did not develop clinical signs of prion disease. In prion-inoculated mice, clinical signs of disease were similar in both α-synuclein transgenic and nontransgenic mice. However, we found a modest but significant acceleration in the time to terminal prion disease in mice having α-synuclein aggregates as compared with nontransgenic mice, with incubation periods of 151 ± 3 d as compared with 162 ± 4 d, respectively (p = 0.02, log-rank test) ().
Figure 1. Survival period of prion-infected α-synuclein transgenic and nontransgenic control mice. (A) In mNS-inoculated mice, survival periods were modestly, but significantly accelerated in mice having α-synuclein aggregates. (B) In contrast, RML-infected mice showed similar survival periods in α-synuclein transgenic and non-transgenic mice.
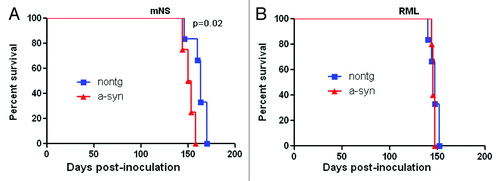
To investigate the possibility of a strain effect in a potential interaction of prions and α-synuclein, we inoculated mice having α-synuclein aggregates and controls with a second strain known as Rocky Mountain Laboratory (RML) mouse-adapted prions. In contrast to results with the mNS prions, α-synuclein mice inoculated with RML mouse prions showed no difference in the prion disease incubation period as compared with control mice ().
Inoculation with mNS or RML prions amplifies α-synuclein pathology in transgenic mice
To evaluate the effects of the infectious prions on α-synuclein aggregate morphology and severity, immunocytochemical analysis with a human α-synuclein antibody was performed. As expected, in the mock-inoculated tg mice there were abundant α-synuclein aggregates in the neuropil and neuronal cell bodies in the neocortex, hippocampus, basal ganglia and brainstem (). In contrast, in the tg mice inoculated with the mNS () or RML prions () there was more abundant accumulation of α-synuclein aggregates in the neuropil () and neuronal cell bodies (). Moreover, there was the presence of α-synuclein immunoreactive plaque-like structures distributed in the neocortex, hippocampus and in the basal ganglia (). Labeled with an antibody against total α-synuclein, these lesions were 30–60 µm in diameter and displayed the presence of abundant peri-neuronal and peri-neuritic α-synuclein aggregates in the mice inoculated with RML and mNS prions (). In association with the increased α-synuclein immunostaining in the neuropil, there were dystrophic neurites of 2–4 µm in diameter (). Comparable dystrophic neuritic profiles were identified with an antibody against phosphorylated α-synuclein (p-Ser129) (). illustrates the plaque-like lesions in the basal ganglia, and the lesions observed in the neocortex and hippocampus had a similar structure ().
Figure 2. Infectious prions promote increased α-synuclein immunoreactivity. (A) α-synuclein aggregates typically deposit within neurons (labeled “n”), shown here in the frontal cortex and hippocampus, and in the neuropil (granular staining) as seen in the mock control. (B) RML prion infection. (C) mNS prion infection. In prion-infected mice, α-synuclein aggregates were more abundant in neurons and in the neuropil (cortex), and additionally appear as scattered plaque-like structures (labeled pl). Vacuoles due to the prion disease are also evident (labeled v). Scale bar = 20 µm. Computer aided image analysis for the levels of α-synuclein immunoreactivity in the neuropil of the frontal cortex, hippocampus and basal ganglia expressed as corrected optical density. (D) Stereological (dissector method) analysis of the estimated numbers of α-synuclein positive neurons in the frontal cortex, hippocampus and basal ganglia expressed as total counts x 102. *p < 0.05 by one way ANOVA with post hoc Dunnet's when comparing to mock control, n = 5 per group.
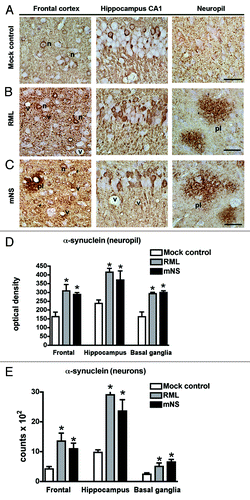
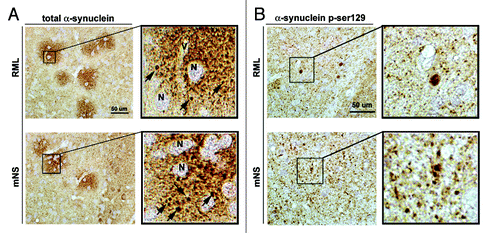
Figure 3. RML and mNS prions results in the formation of plaque-like α-synuclein immunoreactivity structures. All panels are from the basal ganglia. (A) Immunohistochemical analysis with an antibody against total α-synuclein. In RML or mNS prion-inoculated tg mice, abundant α-synuclein aggregates were detected around the neuronal cell bodies (N) and the axonal and dendritic processes. The insets display images at higher power of the α-synuclein plaque-like structures. The arrows indicate dystrophic neurites in the midst of the plaque-like lesions.(B) Immunohistochemical analysis with an antibody against p-ser129 α-synuclein. In RML or mNS prion-inoculated tg mice, abundant α-synuclein aggregates were detected in dystrophic neurites. The insets display images at higher power of α-synuclein affected abnormal axons. Scale bar = 50 µm.
Prion pathology is unaltered by the α-synuclein aggregates
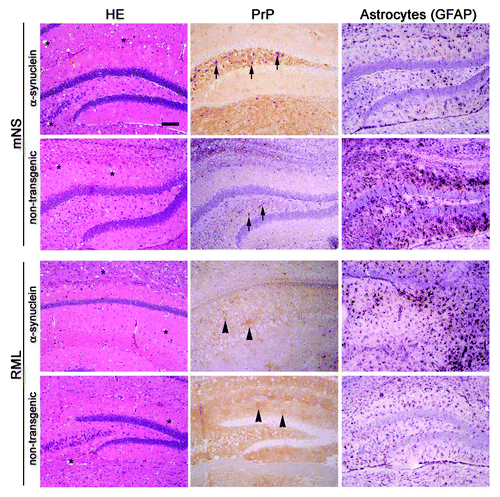
Next we assessed the histopathology of all prion-infected mice to determine whether the prion-induced pathology was altered by the α-synuclein. Infection with mNS causes diffuse aggregates and small 5–10 µm loose plaques whereas RML prions lead to diffuse plaques in brain. Immunohistochemical stains for PrPSc showed that the mNS prion aggregate morphology did not appear to be altered by the presence of α-synuclein. Similar to the mNS strain, the RML aggregate morphology was unchanged by the presence of the α-synuclein aggregates (). Astrogliosis was severe in all prion-infected mice.
Figure 4. Prion-induced histopathology is unmodified by the co-occurrence of α-synuclein aggregates. Brain sections from transgenic (α-synuclein) and non-transgenic mice inoculated with mNS or RML prions show similar spongiform degeneration (labeled as * on HE), PrPSc aggregate morphology, and astrogliosis. mNS prions induce small (2–4 µm), dense, punctuate aggregates (arrows), whereas RML prions induce diffuse, patchy aggregates (arrowheads). Scale bar = 200 µm.
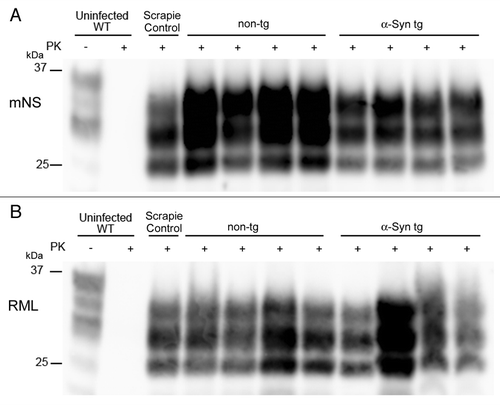
Finally we assessed the PrPSc properties by western blotting. We performed a PK-digest and SDS-PAGE followed by immunoblotting for PrPSc. The glycoform profile and electrophoretic mobility of PrPSc were unaltered by the presence of the α-synuclein (). There was no clear difference in the PrPSc levels in mice with or without α-synuclein aggregates in the brain.
Figure 5. Western blot profiles of PrPSc. A PrP immunoblot for PK-resistant PrPSc reveals similar electrophoretic mobility and glycoform patterns in α-synuclein transgenic and non-transgenic mice inoculated with (A) mNS or (B) RML prions.
Discussion
The present study showed that inoculation of infectious prions into α-synuclein tg mice exacerbated α-synuclein pathology and led to α-synuclein deposits with a neuritic plaque-like appearance. Our study builds on a recent reportCitation24 of prion-infected A53T α-synuclein tg mice and provides new findings. The study by Mougenot and colleagues shows insoluble, phosphorylated α-synuclein in uninoculated and H-BSE inoculated mice at terminal disease (14 mo of age) by western blot and immunohistochemistry. There was no difference in the severity or characteristics of the α-synuclein pathology noted. Here we show a remarkable difference in the α-synuclein pathology in the prion-infected tg mice expressing wildtype α-synuclein. Consistent with our findings, previous studies also suggest that PrPSc promotes the formation of α-synuclein lesions. For example, a previous studyCitation20 showed that α-synuclein immunoreactive deposits accumulate in the brains of patients with sporadic, iatrogenic or variant Creutzfeldt-Jakob disease. In hamsters with experimental scrapie, α-synuclein accumulated near PrPSc deposits but did not co-localize. Similarly α-synuclein has been shown to accumulate in the brain of scrapie-affected sheep and goatsCitation21 and occurred in the hippocampus, a site also affected in our prion-infected α-synuclein mice. Moreover, a recent studyCitation25 reported a case of a 64-y-old woman who developed symptoms of sporadic CJD (sCJD). At autopsy, the brain showed classical methionine/methionine PrPSc type 1 (MM1) sCJD changes, moderate Alzheimer-type pathology, as well as α-synuclein immunoreactive glial cytoplasmic inclusions in the striato-nigral system.
In addition to findings that suggest PrPSc promotes α-synuclein pathology, previous studies have shown that amyloid β in Alzheimer disease accelerates α-synuclein pathologyCitation3 in double tg mouse models and in patients with combined Alzheimer and Parkinson disease.Citation4 Similarly previous reports have shown that Alzheimer disease and CJD can co-exist.Citation26,Citation27 Together these studies suggest that PrPSc promotes α-synuclein aggregation and worsens α-synuclein pathology, and support the notion that misfolded proteins from different neurodegenerative disorders exacerbate disease.
Do preexisting α-synuclein aggregates modify prion disease progression? In our study, prion pathology was unmodified by the overexpression of α-synuclein. However, with the mNS prion strain there was a modest but significant acceleration in the time to terminal prion disease in mice having α-synuclein aggregates as compared with non-tg mice. A recent report showed that tg mice expressing human A53T α-synuclein developed accelerated prion disease compared with non-tg controls when inoculated with scrapie, classic BSE, or H-type BSE.Citation24 The histologic and biochemical features of prion disease were unmodified by the A53T α-synuclein, which was consistent with our findings. The acceleration in this model was greater than the modest effects observed in our study. Such differences may be due to the prion strains used and to the α-synuclein tg mouse model used. Our mThy1- α-synuclein tg mouse model expresses wild type human α-synuclein rather than the mutant type,Citation28 which is known to be more aggressive.Citation29,Citation30 Further supporting the concept that α-synuclein does not worsen the prion pathology, a recent study showed that the severity and progression of prion disease was the same in the absence of α-synuclein.Citation31 Accumulation of PrPSc, synaptic loss and the behavioral deficits associated with the ME7-agent was the same in wildtype and α-synuclein knock out mice.
In conclusion, we showed that infectious prions enhance α-synuclein pathology but not the converse. For future studies, a familial model of prion disease and Parkinson disease would be interesting since α-synuclein and PrP transit through the ERCitation32-Citation34 and are present in lipid rafts.Citation35,Citation36 Important questions that remain to be answered are whether the misfolded PrPSc and α-synuclein interact in the same intracellular compartment and whether the interactions are indirect.
Methods
α-synuclein transgenic mice
All animal procedures were performed in accordance with the protocols approved by the animal care use committee at UCSD following National Institutes of Health guidelines for the humane treatment of animals. All mice were kept in a normal light/dark cycle (12 h light/12 h dark) and had free access to food and water. Mice overexpressing α-synuclein under a Thy-1 promoter non-tg littermate controls (C57/BL6 background) were used. The α-synuclein tgCitation28,Citation37 were previously shown to develop extensive age dependent accumulation of α-synuclein pathology in the neocortex, hippocampus, basal ganglia and brain stem nuclei accompanied by behavioral deficits.Citation38
Prion inoculations
Ten month old α-synuclein tg or non-tg littermate control mice (groups of n = 4–6) were intracerebrally inoculated into the left parietal cortex with 30 µl of brain homogenate containing RML or mNS mouse-adapted sheep scrapie prions. Uninfected brain homogenate was inoculated into the same mouse genotypes as a negative control. Mice were monitored three times weekly, and TSE was diagnosed according to clinical criteria including ataxia, kyphosis, stiff tail, hind leg clasp, and hind leg paresis. Mice were sacrificed at the onset of terminal disease or by approximately 185 d post-inoculation, and incubation period was calculated from the day of inoculation to the day of terminal clinical disease. Mice were maintained under specific pathogen-free conditions.
Histopathology and immunohistochemical analysis
Two-micrometer-thick sections were cut onto positively charged silanized glass slides and stained with hematoxylin and eosin, or immunostained using antibodies for PrP (SAF84) or GFAP for astrocytes. For PrP staining, sections were deparaffinized and incubated for 5 min in 88% formic acid, then washed in water for 5 min, treated with 5 µg/ml of proteinase-K, and washed in water for 5 min. Sections were then autoclaved in citrate buffer (pH 6), cooled for 3 min, and washed in distilled water for 2 min. Immunohistochemical stains were performed using the TSA Plus DNP kit (PerkinElmer). Sections were blocked and incubated with anti-PrP SAF-84 (SPI bio; 1:400) for 45 min followed by anti-mouse HRP (Jackson Immunolabs; 1:500) for 30 min. Slides were then incubated with anti-DNP-HRP (PerkinElmer, 1:100) for 30 min, followed by 6 min incubation with DAB. Sections were counterstained with hematoxylin. For detection of α-synuclein paraffin sections were incubated with a rabbit polyclonal antibody against total α-synuclein (Millipore; 1:500) or a mouse monoclonal against pSer129 α-synuclein (Wako, 1:150).
Computer aided image analysis
The α-synuclein immunolabeled blind coded sections were imaged with a digital bright field photomicroscope (Olympus BX41). Captured images (10 per section) from the mock control and prion inoculated α-synuclein tg mice were delineated and threshold defined and analyzed with the Image Pro-Plus software to determine the corrected optical density value. An additional set of sections counterstained with cresyl violet were analyzed by the dissector method using the Stereo-investigator system to estimate the total number of α-synuclein immunostained neurons in the frontal cortex, hippocampus and basal ganglia.
Western blotting for PrPSc in brain
Samples were electrophoresed in 10% Bis-Tris SDS-PAGE gels (Invitrogen), transferred onto a nitrocellulose membrane, and PrP detected using the anti-PrP primary antibody POM1 (epitope in the globular domain, amino acids 121–231, a kind gift from Dr. Adriano Aguzzi)Citation39 and an HRP-conjugated anti-mouse IgG secondary antibody. The blots were developed using a chemiluminescent substrate (ECL detection Kit, Pierce) and visualized on a Fuji LAS 4000 imager. Quantification was performed using Multigauge V3 software (Fujifilm).
Acknowledgments
We are grateful to Mona Farahi and the animal caretakers for technical assistance. This study was supported by the US. National Institutes of Health [NS055116; NS069566; U54AI065359 (CJS) and AG18440; AG022074; NS057096 (EM)].
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Forman MS, Trojanowski JQ, Lee VM. Neurodegenerative diseases: a decade of discoveries paves the way for therapeutic breakthroughs. Nat Med 2004; 10:1055 - 63; http://dx.doi.org/10.1038/nm1113; PMID: 15459709
- Lansbury PT, Lashuel HA. A century-old debate on protein aggregation and neurodegeneration enters the clinic. Nature 2006; 443:774 - 9; http://dx.doi.org/10.1038/nature05290; PMID: 17051203
- Masliah E, Rockenstein E, Veinbergs I, Sagara Y, Mallory M, Hashimoto M, et al. beta-amyloid peptides enhance alpha-synuclein accumulation and neuronal deficits in a transgenic mouse model linking Alzheimer’s disease and Parkinson’s disease. Proc Natl Acad Sci U S A 2001; 98:12245 - 50; http://dx.doi.org/10.1073/pnas.211412398; PMID: 11572944
- Tsigelny IF, Crews L, Desplats P, Shaked GM, Sharikov Y, Mizuno H, et al. Mechanisms of hybrid oligomer formation in the pathogenesis of combined Alzheimer’s and Parkinson’s diseases. PLoS One 2008; 3:e3135; http://dx.doi.org/10.1371/journal.pone.0003135; PMID: 18769546
- Small SA, Duff K. Linking Abeta and tau in late-onset Alzheimer’s disease: a dual pathway hypothesis. Neuron 2008; 60:534 - 42; http://dx.doi.org/10.1016/j.neuron.2008.11.007; PMID: 19038212
- Head MW, Lowrie S, Chohan G, Knight R, Scoones DJ, Ironside JW. Variably protease-sensitive prionopathy in a PRNP codon 129 heterozygous UK patient with co-existing tau, α synuclein and Aβ pathology. Acta Neuropathol 2010; 120:821 - 3; http://dx.doi.org/10.1007/s00401-010-0766-y; PMID: 21046409
- Harper JD, Lansbury PT Jr.. Models of amyloid seeding in Alzheimer’s disease and scrapie: mechanistic truths and physiological consequences of the time-dependent solubility of amyloid proteins. Annu Rev Biochem 1997; 66:385 - 407; http://dx.doi.org/10.1146/annurev.biochem.66.1.385; PMID: 9242912
- Giasson BI, Forman MS, Higuchi M, Golbe LI, Graves CL, Kotzbauer PT, et al. Initiation and synergistic fibrillization of tau and alpha-synuclein. Science 2003; 300:636 - 40; http://dx.doi.org/10.1126/science.1082324; PMID: 12714745
- Schmidt ML, Martin JA, Lee VM, Trojanowski JQ. Convergence of Lewy bodies and neurofibrillary tangles in amygdala neurons of Alzheimer’s disease and Lewy body disorders. Acta Neuropathol 1996; 91:475 - 81; http://dx.doi.org/10.1007/s004010050454; PMID: 8740227
- Marui W, Iseki E, Uéda K, Kosaka K. Occurrence of human alpha-synuclein immunoreactive neurons with neurofibrillary tangle formation in the limbic areas of patients with Alzheimer’s disease. J Neurol Sci 2000; 174:81 - 4; http://dx.doi.org/10.1016/S0022-510X(99)00327-5; PMID: 10727692
- Morales R, Estrada LD, Diaz-Espinoza R, Morales-Scheihing D, Jara MC, Castilla J, et al. Molecular cross talk between misfolded proteins in animal models of Alzheimer’s and prion diseases. J Neurosci 2010; 30:4528 - 35; http://dx.doi.org/10.1523/JNEUROSCI.5924-09.2010; PMID: 20357103
- Aguzzi A, Calella AM. Prions: protein aggregation and infectious diseases. Physiol Rev 2009; 89:1105 - 52; http://dx.doi.org/10.1152/physrev.00006.2009; PMID: 19789378
- Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science 1982; 216:136 - 44; http://dx.doi.org/10.1126/science.6801762; PMID: 6801762
- Hadlow WJ. Neuropathology and the scrapie-kuru connection. Brain Pathol 1995; 5:27 - 31; http://dx.doi.org/10.1111/j.1750-3639.1995.tb00574.x; PMID: 7767488
- Lashuel HA, Petre BM, Wall J, Simon M, Nowak RJ, Walz T, et al. Alpha-synuclein, especially the Parkinson’s disease-associated mutants, forms pore-like annular and tubular protofibrils. J Mol Biol 2002; 322:1089 - 102; http://dx.doi.org/10.1016/S0022-2836(02)00735-0; PMID: 12367530
- Winner B, Jappelli R, Maji SK, Desplats PA, Boyer L, Aigner S, et al. In vivo demonstration that alpha-synuclein oligomers are toxic. Proc Natl Acad Sci U S A 2011; 108:4194 - 9; http://dx.doi.org/10.1073/pnas.1100976108; PMID: 21325059
- Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L, et al. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci U S A 2009; 106:13010 - 5; http://dx.doi.org/10.1073/pnas.0903691106; PMID: 19651612
- Lee HJ, Suk JE, Patrick C, Bae EJ, Cho JH, Rho S, et al. Direct transfer of alpha-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J Biol Chem 2010; 285:9262 - 72; http://dx.doi.org/10.1074/jbc.M109.081125; PMID: 20071342
- Lee SJ, Desplats P, Sigurdson C, Tsigelny I, Masliah E. Cell-to-cell transmission of non-prion protein aggregates. Nat Rev Neurol 2010; 6:702 - 6; http://dx.doi.org/10.1038/nrneurol.2010.145; PMID: 21045796
- Haïk S, Privat N, Adjou KT, Sazdovitch V, Dormont D, Duyckaerts C, et al. Alpha-synuclein-immunoreactive deposits in human and animal prion diseases. Acta Neuropathol 2002; 103:516 - 20; http://dx.doi.org/10.1007/s00401-001-0499-z; PMID: 11935269
- Adjou KT, Allix S, Ouidja MO, Backer S, Couquet C, Cornuejols MJ, et al. Alpha-synuclein accumulates in the brain of scrapie-affected sheep and goats. J Comp Pathol 2007; 137:78 - 81; http://dx.doi.org/10.1016/j.jcpa.2007.03.007; PMID: 17544436
- Herms J, Tings T, Gall S, Madlung A, Giese A, Siebert H, et al. Evidence of presynaptic location and function of the prion protein. J Neurosci 1999; 19:8866 - 75; PMID: 10516306
- Iwai A, Masliah E, Yoshimoto M, Ge N, Flanagan L, de Silva HA, et al. The precursor protein of non-A beta component of Alzheimer’s disease amyloid is a presynaptic protein of the central nervous system. Neuron 1995; 14:467 - 75; http://dx.doi.org/10.1016/0896-6273(95)90302-X; PMID: 7857654
- Mougenot AL, Bencsik A, Nicot S, Vulin J, Morignat E, Verchère J, et al. Transmission of prion strains in a transgenic mouse model overexpressing human A53T mutated α-synuclein. J Neuropathol Exp Neurol 2011; 70:377 - 85; http://dx.doi.org/10.1097/NEN.0b013e318217d95f; PMID: 21487306
- Rodriguez-Diehl R, Rey MJ, Gironell A, Martinez-Saez E, Ferrer I, Sanchez-Valle R, et al. “Preclinical” MSA in definite Creutzfeldt-Jakob disease. Neuropathology.
- Yoshida H, Terada S, Ishizu H, Ikeda K, Hayabara T, Ikeda K, et al. An autopsy case of Creutzfeldt-Jakob disease with a V180I mutation of the PrP gene and Alzheimer-type pathology. Neuropathology 2010; 30:159 - 64; http://dx.doi.org/10.1111/j.1440-1789.2009.01048.x; PMID: 19703264
- Debatin L, Streffer J, Geissen M, Matschke J, Aguzzi A, Glatzel M. Association between deposition of beta-amyloid and pathological prion protein in sporadic Creutzfeldt-Jakob disease. Neurodegener Dis 2008; 5:347 - 54; http://dx.doi.org/10.1159/000121389; PMID: 18349519
- Rockenstein E, Mallory M, Hashimoto M, Song D, Shults CW, Lang I, et al. Differential neuropathological alterations in transgenic mice expressing alpha-synuclein from the platelet-derived growth factor and Thy-1 promoters. J Neurosci Res 2002; 68:568 - 78; http://dx.doi.org/10.1002/jnr.10231; PMID: 12111846
- Conway KA, Harper JD, Lansbury PT. Accelerated in vitro fibril formation by a mutant alpha-synuclein linked to early-onset Parkinson disease. Nat Med 1998; 4:1318 - 20; http://dx.doi.org/10.1038/3311; PMID: 9809558
- Lim Y, Kehm VM, Li C, Trojanowski JQ, Lee VM. Forebrain overexpression of alpha-synuclein leads to early postnatal hippocampal neuron loss and synaptic disruption. Exp Neurol 2010; 221:86 - 97; http://dx.doi.org/10.1016/j.expneurol.2009.10.005; PMID: 19833127
- Asuni AA, Hilton K, Siskova Z, Lunnon K, Reynolds R, Perry VH, et al. Alpha-synuclein deficiency in the C57BL/6JOlaHsd strain does not modify disease progression in the ME7-model of prion disease. Neuroscience 2010; 165:662 - 74; http://dx.doi.org/10.1016/j.neuroscience.2009.10.047; PMID: 19879926
- Lopez CD, Yost CS, Prusiner SB, Myers RM, Lingappa VR. Unusual topogenic sequence directs prion protein biogenesis. Science 1990; 248:226 - 9; http://dx.doi.org/10.1126/science.1970195; PMID: 1970195
- Hegde RS, Mastrianni JA, Scott MR, DeFea KA, Tremblay P, Torchia M, et al. A transmembrane form of the prion protein in neurodegenerative disease. Science 1998; 279:827 - 34; http://dx.doi.org/10.1126/science.279.5352.827; PMID: 9452375
- Jiang P, Gan M, Ebrahim AS, Lin WL, Melrose HL, Yen SH. ER stress response plays an important role in aggregation of α-synuclein. Mol Neurodegener 2010; 5:56; http://dx.doi.org/10.1186/1750-1326-5-56; PMID: 21144044
- Baron GS, Wehrly K, Dorward DW, Chesebro B, Caughey B. Conversion of raft associated prion protein to the protease-resistant state requires insertion of PrP-res (PrP(Sc)) into contiguous membranes. EMBO J 2002; 21:1031 - 40; http://dx.doi.org/10.1093/emboj/21.5.1031; PMID: 11867531
- Fortin DL, Troyer MD, Nakamura K, Kubo S, Anthony MD, Edwards RH. Lipid rafts mediate the synaptic localization of alpha-synuclein. J Neurosci 2004; 24:6715 - 23; http://dx.doi.org/10.1523/JNEUROSCI.1594-04.2004; PMID: 15282274
- Masliah E, Rockenstein E. Genetically altered transgenic models of Alzheimer’s disease. J Neural Transm Suppl 2000; 59:175 - 83; PMID: 10961430
- Magen I, Chesselet MF. Genetic mouse models of Parkinson’s disease The state of the art. Prog Brain Res 2010; 184:53 - 87; http://dx.doi.org/10.1016/S0079-6123(10)84004-X; PMID: 20887870
- Polymenidou M, Moos R, Scott M, Sigurdson C, Shi YZ, Yajima B, et al. The POM monoclonals: a comprehensive set of antibodies to non-overlapping prion protein epitopes. PLoS One 2008; 3:e3872; http://dx.doi.org/10.1371/journal.pone.0003872; PMID: 19060956