Abstract
Aggregation-prone proteins associated with neurodegenerative disease, such as α synuclein and β amyloid, now appear to share key prion-like features with mammalian prion protein, such as the ability to recruit normal proteins to aggregates and to translocate between neurons. These features may shed light on the genesis of stereotyped lesion development patterns in conditions such as Alzheimer disease and Lewy Body dementia. We discuss the qualifications of tau protein as a possible “prionoid” mediator of lesion spread based on recent characterizations of the secretion, uptake and transneuronal transfer of human tau isoforms in a variety of tauopathy models, and in human patients. In particular, we consider (1) the possibility that prionoid behavior of misprocessed tau in neurodegenerative disease may involve other aggregation-prone proteins, including PrP itself, and (2) whether “prionlike” tau lesion propagation might include mechanisms other than protein-protein templating.
Prions and TSEs as a General Model for Neurodegenerative Disease
The formation of abnormal protein aggregates provides a common element in the pathogenesis of the vast majority of neurodegenerative diseases in humans, including Alzheimer Disease (AD), non-AD tauopathies, α synucleinopathies such as Lewy Body dementia, transmissible spongiform encephalopathies (TSEs), trinucleotide repeat diseases and amyotrophic lateral sclerosis. Since aggregate formation is generally considered (and has been modeled) as a cell-autonomous process, investigations into the pathogenesis of most neurodegenerative diseases have until very recently focused on cytotoxicity mechanisms associated with aggregate formation. As a consequence, intercellular aspects of most neurodegenerative diseases have been relatively neglected until very recently, and remain poorly understood. An exception to this pattern has been the study of TSEs, in which the consequences of prion protein (PrP) misfolding were addressed at the organismal level years before meaningful studies of cellular mechanisms could be attempted. This is because the transmissability and bizarre epidemiology of these diseases made a basic understanding of the transmission mechanism prerequisite to any meaningful studies of TSE cytopathogenesis. However, during the 1980s and 1990s, PrP was progressively established as the necessary and sufficient agent for TSE transmission via the promulgation, systematic testing and validation of the “Prion Hypothesis” Citation1-Citation3. Since then, the study of PrP biology and pathobiology at the molecular, cellular and organismal levels has led to a deepening appreciation of the subtlety with which specific misfolded conformers of PrP can reproduce and propagate disease-specific properties that define specific TSEs. Investigators have recently begun to use PrP propagation in TSEs as a model to address interneuronal aspects of non-prion neurodegenerative diseases such as AD, non-AD tauopathies and Lewy Body disease by focusing on two intriguing features common to these conditions: (1) the sequential involvement of synaptically connected parts of the brain with disease progressionCitation4-Citation7 and (2) synergistic interactions between α synuclein (Asyn), β amyloid (Abeta), tau and PrP, both in cellular disease modelsCitation8,Citation9 and in the distribution of plaques and neurofibrillary lesions in TSE, Lewy Body disease and AD neuropathology.Citation10-Citation12 This has raised the question of whether aggregation-prone proteins such as Asyn, Abeta and most recently tau,Citation13 that have established roles in neurodegenerative disease cytopathogenesis, may also participate in interneuronal lesion propagation.Citation14
The prion-like characteristics of Asyn and Abeta have been reviewed extensively elsewhereCitation13,Citation15-Citation18; this review will focus on the qualifications of tau as a prion—especially in the context of recent findings from our lab and elsewhere suggesting that tau secretion and interneuronal transfer may play a material role in the biogenesis of CSF tau and lesion propagation in AD. Here we will consider whether recent studies demonstrating tau secretion,Citation19-Citation23 uptake,Citation19,Citation24 interneuronal transfer,Citation19,Citation25,Citation26 toxicity,Citation27 and the modulation of these processes by tau aggregation stateCitation24-Citation27 provide sufficient reason to add tau to the growing list of “prionoid” proteins. One of the more interesting consequences of considering the “prionoid” characteristics of tau is that the multiplicity of toxic pathways exhibited by tau highlights the need for re-evaluating basic definitions of “prionoid” and “prionlike,” as is discussed further below.
Membership in the Prion Club is Still Limited to its Founder – PrP
It is important to start by noting that the Prion Club still has only one undisputed member according to the strict “classic” membership criteria laid out 30 y ago in the Prion Hypothesis. This exclusivity is largely due to the controversial nature of protein-only disease transmission at the time and the intense scrutiny that it has received since. The “Prion Hypothesis” of protein–only TSE transmission proposed by Prusiner on the strength of his workCitation1,Citation2 and that of othersCitation28,Citation29 in the early 1980s needed to both exclude a polynucleotide-based mechanism of infectivity and explain the key peculiarities of TSE transmission known at the time. It describes the experimental criteria to be met in demonstrating the transfer and recreation of a complete infective unit between organisms via a purely proteinaceous infectious agent (the “prion”). According to this hypothesis, the minimal infective unit is a protein that a) carries with it the information necessary to convert a resident non-toxic protein into a complete copy of the toxic prion and b) uses endogenous biological mechanisms of the recipient organism so as to permit the infection of additional host organisms. More recent investigations have implicated templated misfolding of PrPC in recipient cells by exogenous PrPSc imported via various mechanisms as the actual propagation mechanism for PrPSc-mediated TSEs, However, this mechanism is not stipulated as a limiting criterion in the Prion Hypothesis. It should also be noted that because the Prion Hypothesis was formulated to account for the peculiar epidemiology of TSEs in mammals, a candidate “prion” also implicitly needs to meet an additional requirement imposed by the multicellular nature of the host —i.e., the ability to propagate between postmitotic cells (such as neurons) within the host organism. Although several prionlike yeast proteinsCitation30-Citation32 have provided invaluable insights into the pathogenesis of TSEs at the cellular level, this latter requirement appears to exclude yeast prions from full membership by virtue of the unicellular nature of their natural hosts, although they meet the other formal criteria for Prion Club membership.Citation32,Citation33 Yeast “prions” share certain general structural features (i.e., enrichment in N, Q, P and related amino acids) with mammalian PrP, but have little specific sequence homology to it.Citation34 They have thus been particularly useful in distinguishing generally applicable prion characteristics from those that are PrP-specific and in validating the Prion Hypothesis as a concept. However, a complete understanding of TSE etiology and pathogenesis ultimately depends on elucidating the link between “normal” and pathological functions of PrP.
Insights into “Prionoid” Lesion Propagation Mechanisms from TSE Models
Cellular and transgenic PrP-based models have been successfully used to reproduce subtle characteristics of TSEs at the molecular (templated alterations in protein conformation), cellular (secretion, misprocessing) and organism (infectivity) levels. A prime example of this is the maintenance of strain-specific clinical and neuropathological differences between individual TSEs, which now appear to be associated with specific misfolded PrP conformations,Citation35-Citation37 that are maintained even when the infection source is purely sporadic.Citation38 We now know that subtle differences in PrPSc tertiary structure between disease strains are modulated not only by polymorphisms at specific key loci in the PrP sequence (e.g., position 129 in familial TSEs induced by the D178N mutation) but also by polymorphisms elsewhere in the molecule associated with expansion of the octapeptide repeat region.Citation39 It is also becoming apparent that the propagation of misfolded PrP through the CNS of an infected individual and between different individuals occurs on a different timetable than the cytotoxic consequences of PrP misfolding,Citation40,Citation41 with propagation to accessible vulnerable tissues appearing to precede the development of cytotoxicity.Citation40 This is consistent with the observed separation of templating, propagation and toxicity-mediating domains of PrP, and may shed light on the criteria for prion-like changes in other proteins with analogous structural and functional motifs. However, it also highlights the relative lack of information about the actual propagation mechanism of PrPSc between cells as opposed to the detailed characterizations available for the templating mechanism itself.
PrPC is primarily localized to the plasma membrane and membrane-associated trafficking vesiclesCitation42 and plays a role in membrane-associated cellular functions such as process outgrowth.Citation43,Citation44 Transformation of PrPC to PrPSc occurs in close association with early endosome formation, thereby linking membrane trafficking with TSE cytopathogenesis and PrPSc propagation.Citation45 The GPI anchor mediated membrane link of PrP appears to be intimately involved with PrPSc propagation in particular, as even subtle modifications result in major alterations in propagation efficiency and pattern in transgenics.Citation46-Citation50 The role of PrPC in synaptic plasticity and signal transductionCitation51 in particular may account for unique PrP characteristics (such as the subtlety of PrP templating in TSEs) and could explain (at least partly) why PrP is the only identified mediator of protein-only disease transmission among mammals. A possible analog for a prionlike normal PrP function is provided by a synaptic plasticity protein (cytoplasmic polyadenylation element binding protein or CPEB) in the marine snail Aplysia. The Aplysia CPEB has multiple highly stable conformations that confer stability to associated memory states and appear to involve localized templating interactions (model)Citation52 that are highly reminiscent of abnormal PrPSc function in TSEsCitation53. Expression of the Q/N rich N-terminal domain of CPEB in yeast confers heritable conformation changes, thus making CPEB a prion in the same sense as yeast prions are.Citation54
“There Goes the Neighborhood”- The Case for Auxiliary Membership for “Prionoid” Proteins
The early brain inoculate studies in chimpanzees conducted by Gadjusek in the 1960sCitation29 demonstrated the unique transmissability of PrP-based TSEs relative to other neurodegenerative conditions, including AD and Parkinson Disease. However, it is the interneuronal transfer of PrPSc, rather than interorganismal transmissability per se that has attracted attention as a potential general model of non-TSE neurodegenerative disease pathogenesis. Recent studies of other proteins associated with aggregation-driven toxicity in neurodegenerative conditions have raised the question of whether an associated, less exclusive “prionoid”Citation16 status should be designated in the Prion Club for proteins capable of mediating the interneuronal propagation of neurodegeneration-inducing toxicity rather than that of transmission between individuals. This would provide an attractive hypothetical framework for considering the mechanisms responsible for the stereotyped progression of neurofibrillary lesions through the brain in ADCitation5,Citation7 Parkinson diseaseCitation6 and other tauopathies.Citation8 Of the 3 aggregation-prone proteins (e.g., Asyn, Abeta and tau) involved in the majority of human neurodegenerative conditions, Abeta and Asyn have received the most attention, and explicit cases have been made for their designation as prionlike agents in the pathogenesis of AD and Lewy Body dementias respectively.Citation55,Citation56 Both proteins are secreted from neurons,Citation57,Citation58 where they induce localized toxicity via either uptakeCitation59,Citation60 or receptor-mediated mechanisms.Citation61 Some in vivo evidence for aggregation-mediated lesion propagation exists for both Abeta and Asyn; intraperitoneal injection of Abeta into mice transgenic for familial AD mutations in amyloid precursor protein have been shown to accelerate the onset of senile plaque formationCitation56 whereas long-term fetal grafts into Parkinson Disease patients have exhibited Lewy Body pathology that can only be plausibly accounted for by a lesion spreading mechanism.Citation55 It is worth noting that other neurodegenerative conditions driven by aggregation-prone proteins or protein sequences also share “prionoid” properties. Examples include SOD1, TDP-43, and polyQ containing proteins, with the latter being particularly interesting in the context of the high QN content of “consensus” regions identified in various yeast prions.Citation35 The degree to which key aggregation-prone proteins discussed satisfy Prion Club requirements is summarized in .
The Case for Tau as a Member in the Expanded Prion Club
Unlike Asyn and Abeta, tau is normally a cytosolic protein, expressed primarily in neurons and glia,Citation62 with a significant role in the modulation of microtubule (MT) stability,Citation63,Citation64 especially in axons, where it appears to play a role in establishing neuronal polarity and axonal identity.Citation65 While tau has been widely supposed to play an exclusively cytosolic/cytoskeletal role both normally and when misprocessed to form neurofibrillary aggregates, there has been increasing evidence that tau interacts with plasma membrane elements and becomes involved in membrane-associated mechanisms, including secretion, ever since the mid-1990s, when Lee and coworkers demonstrated that the tau N-terminal “projection” domain interacts with lipid raft-associated non receptor tyrosine kinases.Citation66,Citation67 Tau has since been shown to have well defined roles in axonal outgrowth, guidance and myelination that involve tyrosine kinases (e.g., fyn) involved in signal transduction, endocytosis and vesicle trafficking,Citation68-Citation70 and which appear also to be involved in tau secretion.Citation22 However, while tau resembles PrP and Asyn in having a tertiary structure that is delicately balanced between an extended normal conformation and one prone to self-aggregation,Citation15 it now appears that there are toxicity pathways associated with tau misprocessing that are not directly associated with classic “prionlike” templated misfolding. This raises fundamental questions about the meaning of “prionlike” that must be addressed when considering the Club membership of tau.
Tau Secretion and Toxicity in Models
Despite its cytosolic reputation, the possible involvement of secretion in disease-associated tau misprocessing was evident in the first successful “tauopathy” model,Citation71-Citation73 in which human tau expressed cell autonomously in identified lamprey central neurons (ABCs) was released in the vicinity of degenerating dendrites. Further studies in this system established that tau secretion was fully separable from tau-induced toxicity by either the application of small molecule inhibitors of tau aggregation,Citation25,Citation74 or by the removal of the MT binding repeat (MTBR)-containing C-terminal domain.Citation19 Tau thus exhibits the full range of possible propagation routes necessary for Prion Club membership, at least in the lamprey model (). In all models in which tau secretion has been characterized, the presence of the N terminus has been essential for secretion, the efficacy of which is modulated by the presence of the alternatively spliced exon 2 insert.Citation20 By contrast, the uptake of tau from the extracellular space does not appear to require the tau N terminus, and rather appears to be linked to the aggregation state of the tau MTBR.Citation24 Extracellular tau toxicity can be either due to tau uptakeCitation24 or via receptor mediated Ca2+ fluxes.Citation75 One of the two identified mechanisms of tau-induced toxicity is associated with MTBR mediated aggregation,Citation76,Citation77 and disease-specific lesion characteristics at the molecularCitation78 cellularCitation79 and intercellular levels of analysis,Citation5-Citation7,Citation80 and thus appears analogous to PrP-style templated misfolding. Tau filament formation is itself catalyzed by interactions with membrane lipidsCitation81 and with perimembranous extracellular matrix moleculesCitation82 as well as via direct templating with other MTBRs,Citation83 suggesting a subtlety reminiscent of PrP-mediated pathogenesis. The other toxicity mechanism is specific to the N terminusCitation61,Citation84-Citation86 and mediates Abeta cytotoxity.Citation87 While N-terminal tau toxicity does not require the MTBRCitation85,Citation86 and thus does not appear to involve templated misfolding, it could in theory propagate tau aggregate-containing lesions by modulating caspase and calpain cleavage of endogenous tau in recipient cells. This would generate both aggregation-prone tau fragments containing the MTBRCitation88,Citation89 and additional MTBR-less N-terminal fragments, whose secretion could mediate further interneuronal lesion propagation ().
Figure 1. Human tau is propagated transsynaptically, to adjacent neurons and to the CSF in the lamprey tauopathy model. (Panel A) Confocal images of a dendrodendritic apposition between 2 different identified lamprey reticulospinal neurons (ABCs) expressing the wild-type (WT) 4R0N human tau isoform in a transverse section through lamprey brain 20 d after plasmid injection. The right image shows the region inset at left. N-terminally tagged tau can be seen in exocytosing profiles (arrows) near the points of dendrodendritic contact (asterisks). Dendrodendritic anastmoses have not previously been described in lamprey reticulospinal neurons. (Panel B) Sonatodendritic (top left) and axonal (bottom left) cross-sectional profiles through ABCs expressing the 4R0N human tau isoform containing the P301L tauopathy-inducing mutation. P301L tau is diffusely secreted from the somata of expressing ABCs (asterisk, top left) crosses the IVth ventricle (IV) and enters adjacent neuronal somata (arrow, top left). Bottom Left: Somatic tau staining (s) can be seeen in a neuron that is postsynaptic to the axonal profile (a) of a tau-expressing ABC. Right image in B shows periventricular regions from the hindbrain of a lamprey that contained P301L-expressing ABCs (not present in section). Note that periventricular tau has moved into and bilaterally within the ventricle (IV, asterisk marks midline), crossed the ependymal cell layer (ep) and entered small neurons (n) on both sides of the brain (arrows). The gradient of immunolabel (inset) suggests that exogenous tau from the ventricle has penetrated brain tissue. (Panel C) Transverse sections through brains of lampreys that were treated with control vehicle (DMSO – leftmost image) and anti tau aggregant (NNI3 - right 3 images) as described in.Citation25 The rightmost section was taken at a point 100 microns rostral to the ABC dendritic field limit, and shows 3R0N tau-positive afferents to expressing ABCs (shown in center-right image) and thus illustrates retrograde transsynaptic transport of human tau.The center-right image shows a tau-expressing ABC and also tau-positive profiles of dendrites from adjacent non-expressing ABCs, suggesting dendrodendritic tau movement. Note that secretion, retrograde and transventricular tau movement are all enhanced by a tau anti-aggregation agent (NNI3), resulting in clearance of tau from overexpressing ABCs and uptake into periventricular ependymal cells (center left image). The section shown in A was immunostained for the N-terminal GFP epitope tag (red) a general tau marker (Tau5 – green) and filamentous actin (phalloidin – blue). The sections shown in B and C were immunolabeled with the human tau specific mAb Tau12 to the tau N terminus (residues 9–18) and revealed using standard HRP/DAB immunohistochemistry. Scale Bars: Panel A: 20 microns, Panel B 50 microns,(left): 100 microns (right), Panel C 100 microns
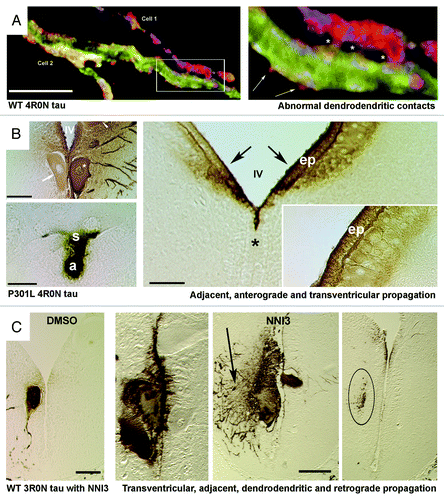
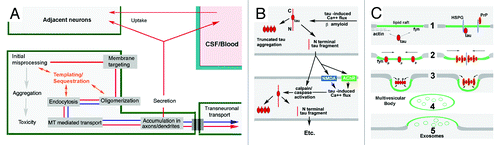
Figure 2. Candidate neuronal and interneuronal pathways of self-replicating tau toxicity and propagation. The generation of PrPSc from resident PrPC occurs via a chaperone-like “templating” in association with mambrane-associated misprocessing events that eventiually result in exosome mediated PrPSc release to the extracellular space and transsynaptic propagation of infective prions to other parts of the CNS. (A and B) Hypothesized templated (A) and non-templated (B) tau lesion propagation pathways consistent with the Prion Hypothesis. (A) Recent studies of tau misprocessing suggest that tau misprocessing may occur via a similar mechanism (shown in red arrows) that permits synergistic interactions with PrP and other “prionoid” proteins such as Asyn and Abeta (shown in blue arrows). Either overexpression or toxicity-induced hyperphosphorylation causes the generation of non-MT associated tau which becomes associated with and phosphorylated by fyn kinase and other elements in the plasma membrane (e.g., PrP, Asyn, Abeta), resulting in the oligomerization, endocytosis and relocalization of tau to pre and postsynaptic sites within the neuron, where it accumulates and causes localized toxicity and exosome-mediated secretion. The ability of tau to selectively sequester other MAPs and bind/oligomerize with other aggregation prone proteins such as Asyn and PrP suggests that template-mediated misfolding might occur in this pathway (orange) (Panel B) Tau lesion propagation might also occur via the generation of toxic tau cleavage fragments by Abeta or tau-induced calpain and caspase activity and their secretion via a separate pathway. N-terminal truncated, MTBR containing, tau fragments exhibit an increased tendency to aggregate.Citation88,Citation89 MTBR- fragments are readily secreted and may induce calpainCitation84/caspaseCitation85 activation in recipient neurons increased resulting in self-regenerating tau lesion propagation. (C) Tau/PrP interactions in exosomal secretion. Two possible ways in which membrane associated tau misprocessing might result in diversion to the exomal secretion pathway. Usually considered cytosolic, non-MT-associated tau can be targeted to lipid raft domains in the plasma membrane by binding ligands such as fyn kinase or subcortical actin (1). Tau is phosphorylated by fyn (or a related Y kinase) and oligomerizes (2), possibly with the aid of polyanionic molecules within (fatty acids) or spanning (heparan sulfate proteoglycans (HSPG)) the membrane, causing raft patching, endocytosis (3) and the formation of multivesicular bodies (4) and exosome release (5). Although the GPI anchoring of PrP should direct it to the exterior of the plasma membrane, the presence of polyanions such as HSPG may bring it into direct contact with tau and permit synergistic co-oligomerization. Other aggregation-prone proteins such as Abeta and/or Asyn may play similar, possibly context-dependent roles with respect to the misfolding and interneuronal propagation of PrP and/or tau.
The consideration of a possible “prionoid” role for tau in lesion propagation is more recent and more tentative than for Abeta and Asyn, despite spatiotemporal lesion development in AD and non-AD tauopathies suggestive of transsynaptic tau movementCitation6,Citation7 and demonstrations of toxicity-associated secretionCitation71,Citation73, interneuronal mobilityCitation25 and active release to the CSF in non-murine tauopathy models (). This is very likely due to unique difficulties of modeling tau toxicity and secretion in more widely used cell culture and murine transgenic tauopathy models. In cultured neurons, overexpression of human tau isoforms has consistently failed to produce tau specific toxicityCitation90 without the application of toxic, broad spectrum inducers of protein phosphorylationCitation91 or the use of hyperaggregating mutants,Citation76 although overexpression at similar levels in situ readily induces tau hyperphosphorylation, fibrillogenesis and neurodegeneration.Citation71-Citation73,Citation92,Citation93 This is presumably due to a dependence of tau-induced toxicity either on terminal differentiation of the neuronal stateCitation94 or on the retention of toxic secreted tau in the perineuronal extracellular space,Citation27,Citation95 neither of which can occur in cell culture. Initial attempts to model human tau induced toxicity in murine transgenics used wild-type tau isoforms that were only mildly overexpressed relative to human tau levels and did not induce neurodegeneration, although they did exhibit somatic accumulation of phosphorylated, non-MT-associated human tau.Citation96,Citation97 Signs of neurofibrillary degeneration were only observed with stronger (5x human) overexpression of wild-type human tau isoforms, and then only in aged animals.Citation92,Citation93 The first widely used mouse model of familial tauopathy was generated using the overexpression of a tauopathy mutant tau isoform (P301L).Citation98 Tau secretion was not identified in any of these models, presumably owing to the difficulty of determining the source of secreted tau against a tau–positive background, a problem that was not encountered in cell-autonomous, non-transgenic models such as the lamprey. This problem was eventually elegantly circumvented in a study that implicitly demonstrated tau-mediated lesion propagation by showing that tau-induced aggregate formation could be propagated over significant distances within the brain of a mouse already transgenic for human tau.Citation26
Despite widespread reservations about the use of overexpression in disease models, the nature of tau-induced toxicity makes it likely that in situ overexpression of human tau faithfully replicates the downstream disease mechanisms (i.e., toxicity and secretion) that drive AD and tauopathy pathogenesis. Externally applied Abeta 1–42 induces tau hyperphosphorylation and MT lossCitation99 as does the presence of familial tauopathy mutations,Citation100,Citation101 effectively raising the cytosolic concentration of non-MT-associated tau (as does tau overexpression) and thus favoring tau secretion. The repeated observation that tau overexpression induces AD-like tau hyperphosphorylation is also consistent with the cytopathological effects of Abeta-mediated tau hyperphosphorylation in AD, and suggests that both hyperphosphorylation and overexpression both result in the accumulation of nonMT-associated tau in somatodendritic locations, albeit by different mechanisms. Tau secretion in the lamprey model occurs preferentially from dendritic loci where non MT-associated tau has accumulated and MTs are being lost prior to the onset of degeneration,Citation22,Citation72,Citation73 suggesting that such tau is both locally toxicCitation27,Citation76 and prone to secretion.Citation19,Citation22 Tau secretion that disposes of intracellular accumulations may be neuroprotective to the neuron expressing tau,Citation25,Citation95 but may also generates extracellular tau that is toxic to other neuronsCitation27,Citation75 The neuroprotective and pro-secretory effect of the anti-tau aggregant NNI3Citation25 may be due to the ability of NNI3 to to block high level tau oligomer/polymer formation,Citation74 which could induce accumulation of lower level oligomers in membrane-associated tau and possibly favor its secretion,Citation102 thereby preventing the intracellular buildup of toxic, non-MT-bound tau.
Secreted, Oligomerized Phosphotau in Human CSF
The recent finding that elevated CSF tau in early AD appears to be secretedCitation21 constitutes the first direct link between tau secretion and human disease pathogenesis and replicates the transventricular propagation of tau that has been demonstrated in the lamprey model () and suggested in recent mouse model studies.Citation23 It also raises novel possible mechanisms for prionoid lesion propagation. For instance, CSF-tau levels are significantly more elevated in AD than in non-AD tauopathies,Citation103 suggesting that Abeta-tau interactions – possibly involving the tau N-terminal projection domainCitation61,Citation85 - might enhance any tau secretion responsible for the genesis of elevated CSF-tau. This is consistent with the overrepresentation of N-terminal tau species in CSF from AD patientsCitation104 and the restriction of significantly elevated CSF tau to AD (relative to most non-AD tauopathies), but is not necessarily indicative of a classical “prionlike” mechanism, since the MTBR of tau is not needed for either tau secretion or Abeta-mediated toxicity.Citation19,Citation85 However, recent findings that exposure to CSF from Parkinson disease (which contains oligomeric Asyn) is cytotoxicCitation105 raise the possibility that elevated CSF tau may play a direct role in AD pathogenesis that may not be present in non-AD tauopathies. The finding that some of the membrane-associated tau in AD may be oligomeric is especially interesting in this context, as it is more consistent with a PrP-like templated toxicity mechanism.Citation21
Is Tau Lesion Spreading due to Synergistic Interactions with PrP?
While improved techniques have overcome most limitations to the infectivity and in vivo propagation of artificial prions,Citation106 this propagation still requires cytosolic and membrane-associated cofactors capable of stabilizing the transient intermediate conformations that occur in PrPC to PrPSc conversion to complete the transformation of PrPC into fully infectious PrPSc Citation107–Citation108. In particular, it is still unclear whether the strain-specific PrP conformations that give rise to specific TSEs require particular cellular contexts (and ligands) to express the disease specific features of any given TSE.Citation46-Citation49 One possible context-dependent ligand that might modulate PrP-mediated disease is tau, which exhibits significant neuropathological and neurochemical abnormalities in at least some TSEs.Citation10 NFT formation is a prominent aspect propagated lesions in Creutzfeld-Jakob Disease (CJD) in as well as in AD, while CSF-tau levels are even higher in CJD than they are in AD, raising the possibility that PrPSc in at least some TSEs may induce tau misprocessing, especially since tau has recently been shown to bind to PrP on a disease-associated basis.Citation109 However, the implications of such interactions are unclear, as a recent mouse model study suggests that knockout of endogenous tau does not modulate conversion of PrPc to the toxic PrPSc form,Citation110 while another study in a cellular overexpression model demonstrated a tau-mediated depletion of endogenous PrP at the plasma membrane that was correlated with PrP toxicity.Citation111 That said, there are numerous similarities between tau and PrP misprocessing pathways that would seem to present ample opportunity for these proteins to interact in ways that would favor their mislocation, aberrant phosphorylation, oligomerization and secretion (see ). Both PrPSc generation from PrPC and membrane-associated events in tau misprocessingCitation20,Citation21 are localized to subcortical actin-rich structures that include early endosomes.Citation42,Citation112,Citation113 Both tau and PrP exhibit disease-associated interactions with the lipid raft-associated tyrosine kinase fyn in endocytotic pathways that characteristically involve the oligomerization of fyn-associated membrane-associated proteinsCitation70,Citation102,Citation114,Citation115, in some cases via substrate.Citation70 This last is of particular interest since both tau and PrP have now been shown to undergo exosome-mediated secretionCitation21,Citation116 in association with disease-associate misfoldingCitation117 and since oligomerization may cause the diversion of a broad range of membrane-associated proteins into the exosomal secretion pathway.Citation118 Moreover, tau and PrP colocalize and interact with common polyanionic ligandsCitation82,Citation119-Citation122 including HSPGs associated with unconventional secretion pathways.Citation123 The relocalization of either tau or PrP with respect to membrane topology via polyanion binding could be important in increasing synergistic interaction between tau and PrPCitation111 and their endocytosis,Citation70,Citation124 hypothetically leading to oligomerization of tau and/or PrP as well as their secretion (). Finally, while most tau is MT-bound and axonally localized, tau is typically disassociated from MTs and relocalized to the soma and dendrites in AD and non-AD tauopathies, and has recently been identified in dendritic spines, where it has a tauopathy mutation-specific effect on synaptic functionCitation125–Citation127. Thus the specific association and modification of tau with/by abnormal ligands may well play a key role in disease-associated tau misprocessing. Similarly, differential processing of PrPSc in various TSEs suggest that TSE disease identity is to some extent dependent on differential interactions between PrP and ligands capable of stabilizing critical conformation intermediate states between PrPC and PrPSc. Candidate ligands (e.g., Asyn, 14–3–3 proteins) have known chaperone-like functions and are known to interact with membrane-associated polyanionsCitation128 as well as both PrP and tau.Citation120,Citation129,Citation130 Conversely, it is also possible that disease-specific elements of tauopathies (e.g., propagation rateCitation131 might be due to different affinities of tau for PrP imposed by disease mutations or combined with isoform specific misprocessing elements. Conformational changes induced in tau by the P301L tauopathy mutation may alter the relationship between tau and specific tau-binding proteins (e.g., AsynCitation132) that are known to have effects on tau conformation and MT-binding.Citation8 This may also account for the disproportionate presence of specific tau isoforms (i.e., exon 2+, exon 10+) in the elevated CSF-tau levels typically present in CJD.Citation133
Summary and Conclusion
In summary, current research in models and very recently human patients suggests that toxic forms of tau can (A) be secreted to ISF and other body fluid compartments (CSF, blood) and be taken up by adjacent neurons and (B) be retrogradely and anterogradely transported between neurons across synapses. Specific secretion pathways have been identified that are modulated by defined regions of the tau molecule. There is also clear evidence that oligomerized tau is itself toxic,Citation134 with extracellularly applied tau being more toxic,Citation27 more susceptible to uptakeCitation24 and able to “seed” new aggregatesCitation60 in an oligomeric state than it is as monomer. Misprocessed (e.g., hyperphosphorylated, oligomerized, mutant) tau shows features that are consistent with templating, including an altered conformationCitation135 the selective sequestration of tau family MAPs by misprocessed tauCitation83 and the multiple filament morphologies encoded by aggregated tau isoforms.Citation78 These characteristics suggest that tau might be capable of disease-specific templating under ideal conditions, some of which may involve poorly understood interactions with other aggregation-prone proteins, including PrP itself. However, it remains possible that tau toxicity transfer could be an important disease mechanism without involving templated misfolding, especially in AD, where established toxicity routes for the known causative agent (β amyloid) include MTBR-independent toxicityCitation61,Citation86 It is still unclear whether the features characterizing tau secretion and toxicity described by our lab and others in various tauopathy models will apply directly to AD and other tauopathies, but the recent association of exosomal tau secretion and elevated CSF-tau in ADCitation21 and the generation of a mouse model for early-stage tau lesion spreadCitation136 suggests that they will have significant clinical relevance. Tau’s credentials for membership in the “prionoid” auxiliary of the Prion club may therefore be said to be currently under review, but appear to be broadly similar to those of A β and asyn, even if only templating-mediated mechanisms are considered.
While the lack of direct evidence for interneuronal as well as inter-organismal transmission for tau in human neurodegenerative disease is likely due to a more limited ability of tau proteins to template and propagate than is true for PrP, it may also reflect the current rudimentary understanding of the role of tau transfer in neurodegenerative disease pathogenesis. Our assessment is further complicated by what is still a relatively poor understanding of the prion propagation mechanism – how PrPSc is actually transferred between neurons - beyond the established ability of PrPSc to move between neurons. While it is clear that PrPSc can be secreted via the exosome pathwayCitation56 in a number of cell types including neuronsCitation137 and it appears likely that this mechanism is operative in gut-transmitted TSEs (such as vCJD)Citation117,Citation138 it is as yet unclear whether this is actually how disease transmission occurs between neurons in TSEs. PrP possesses a “signal” sequence and can be incorporated into extracellular aggregates via the classical secretion pathway via cleavage of its GPI anchor,Citation48 but there is as yet no evidence that this is an important pathway in the cytopathogenesis of TSEs. Finally, the multiplicity of toxicity routes (some of which do not involve templated misfolding) available to tau,Citation86,Citation87 and other “prionlike” protein candidates (e.g., TDP-43)Citation139 raise issues that are central to both tau membership (e.g., are both toxic tau and PrPSc generated and propagated in a common exosomal pathway in TSEs and/or tauopathies?) and a more fundamental question – what does “prionlike” mean? Does it merely require that the original Prion Hypothesis be satisfied in the context of interneuronal lesion transfer, or does it also require that the toxicity and propagation mechanism involve templated protein misfolding, the mechanism now generally accepted as mediating PrP pathogenesis? It is possible to envision a scenario (diagrammed in ) by which neurofibrillary lesions are spread via the secretion of N-terminal tau fragmentsCitation19 that act via amplifying excitotoxicityCitation75,Citation85 in the recipient cell,Citation61,Citation87 especially in AD, where such fragments constitute the majority of CSF-tau species.Citation104 This is of course highly speculative (as is the case with the now much remarked-upon similarities between aggregation-prone proteins such as tau and PrP) but it again raises the possibility that the original concept of protein mediated toxicity transfer may need to be updated.
So in short, the background check on tau is incomplete at the Prion Club, but fraternization between applicants and members has already begun, with possible widespread consequences for the meaning of Club membership.
| Abbreviations: | ||
| PrP | = | prion protein |
| PrPSc | = | misfolded disease-associated PrP |
| PrPC | = | Normal (cellular) PrP |
| TSE | = | transmissible spongiform encephalopathy |
| CJD | = | Creutzfeld Jakob Disease |
| AD | = | Alzheimer’s Disease |
| MTBR | = | microtubule binding repeat |
| Asyn | = | alpha synuclein |
| Abeta | = | beta amyloid peptide |
| CPEB | = | cytoplasmic polyadenylation element binding protein |
| GPI | = | glycophosphoinositol |
| HSPG | = | heparan sulfate proteoglycan |
| MT | = | microtubule |
| CSF | = | cerebrospinal fluid |
| CNS | = | central nervous system |
| 4R0N | = | tau isoform with 4 microtubule binding repeats |
| Exon 2,3 – | = | tau lacking exons 2 and 3 |
References
- Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science 1982; 216:136 - 44; http://dx.doi.org/10.1126/science.6801762; PMID: 6801762
- Prusiner SB, Bolton DC, Groth DF, Bowman KA, Cochran SP, McKinley MP. Further purification and characterization of scrapie prions. Biochemistry 1982; 21:6942 - 50; http://dx.doi.org/10.1021/bi00269a050; PMID: 6818988
- Büeler H, Aguzzi A, Sailer A, Greiner RA, Autenried P, Aguet M, et al. Mice devoid of PrP are resistant to scrapie. Cell 1993; 73:1339 - 47; http://dx.doi.org/10.1016/0092-8674(93)90360-3; PMID: 8100741
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 1991; 82:239 - 59; http://dx.doi.org/10.1007/BF00308809; PMID: 1759558
- Braak H, Ghebremedhin E, Rüb U, Bratzke H, Del Tredici K. Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res 2004; 318:121 - 34; http://dx.doi.org/10.1007/s00441-004-0956-9; PMID: 15338272
- Armstrong RA, Cairns NJ, Lantos PL. What does the study of the spatial patterns of pathological lesions tell us about the pathogenesis of neurodegenerative disorders?. Neuropathology 2001; 21:1 - 12; http://dx.doi.org/10.1046/j.1440-1789.2001.00373.x; PMID: 11304036
- Su JH, Deng G, Cotman CW. Transneuronal degeneration in the spread of Alzheimer’s disease pathology: immunohistochemical evidence for the transmission of tau hyperphosphorylation. Neurobiol Dis 1997; 4:365 - 75; http://dx.doi.org/10.1006/nbdi.1997.0164; PMID: 9440125
- Giasson BI, Forman MS, Higuchi M, Golbe LI, Graves CL, Kotzbauer PT, et al. Initiation and synergistic fibrillization of tau and alpha-synuclein. Science 2003; 300:636 - 40; http://dx.doi.org/10.1126/science.1082324; PMID: 12714745
- Laurén J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 2009; 457:1128 - 32; http://dx.doi.org/10.1038/nature07761; PMID: 19242475
- Giaccone G, Mangieri M, Capobianco R, Limido L, Hauw JJ, Haïk S, et al. Tauopathy in human and experimental variant Creutzfeldt-Jakob disease. Neurobiol Aging 2008; 29:1864 - 73; http://dx.doi.org/10.1016/j.neurobiolaging.2007.04.026; PMID: 17560687
- Clinton LK, Blurton-Jones M, Myczek K, Trojanowski JQ, LaFerla FM. Synergistic Interactions between Abeta, tau, and alpha-synuclein: acceleration of neuropathology and cognitive decline. J Neurosci 2010; 30:7281 - 9; http://dx.doi.org/10.1523/JNEUROSCI.0490-10.2010; PMID: 20505094
- Haïk S, Privat N, Adjou KT, Sazdovitch V, Dormont D, Duyckaerts C, et al. Alpha-synuclein-immunoreactive deposits in human and animal prion diseases. Acta Neuropathol 2002; 103:516 - 20; http://dx.doi.org/10.1007/s00401-001-0499-z; PMID: 11935269
- Novak P, Prcina M, Kontsekova E. Tauons and prions: infamous cousins?. J Alzheimers Dis 2011; 26:413 - 30; PMID: 21694453
- Cushman M, Johnson BS, King OD, Gitler AD, Shorter J. Prion-like disorders: blurring the divide between transmissibility and infectivity. J Cell Sci 2010; 123:1191 - 201; http://dx.doi.org/10.1242/jcs.051672; PMID: 20356930
- Hall GF. What is the common link between protein aggregation and interneuronal lesion propagation in neurodegenerative disease? in “Neurodegenerative Diseases - Processes, Prevention, Protection and Monitoring”,2011 R. Chang ed. InTech pp 1-17.
- Aguzzi A, Rajendran L. The transcellular spread of cytosolic amyloids, prions, and prionoids. Neuron 2009; 64:783 - 90; http://dx.doi.org/10.1016/j.neuron.2009.12.016; PMID: 20064386
- Moreno-Gonzalez I, Soto C. Misfolded protein aggregates: mechanisms, structures and potential for disease transmission. Semin Cell Dev Biol 2011; 22:482 - 7; http://dx.doi.org/10.1016/j.semcdb.2011.04.002; PMID: 21571086
- Brundin P, Melki R, Kopito R. Prion-like transmission of protein aggregates in neurodegenerative diseases. Nat Rev Mol Cell Biol 2010; 11:301 - 7; http://dx.doi.org/10.1038/nrm2873; PMID: 20308987
- Kim W, Lee S, Jung C, Ahmed A, Lee G, Hall GF. Interneuronal transfer of human tau between Lamprey central neurons in situ. J Alzheimers Dis 2010; 19:647 - 64; PMID: 20110609
- Kim W, Lee S, Hall GF. Secretion of human tau fragments resembling CSF-tau in Alzheimer’s disease is modulated by the presence of the exon 2 insert. FEBS Lett 2010; 584:3085 - 8; http://dx.doi.org/10.1016/j.febslet.2010.05.042; PMID: 20553717
- Saman S, Kim W, Raya M, Visnick Y, Miro S, Saman S, et al. Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J Biol Chem 2012; 287:3842 - 9; http://dx.doi.org/10.1074/jbc.M111.277061; PMID: 22057275
- Lee S, Kim W, Li Z, Hall GF. Accumulation of vesicle-associated human tau in distal dendrites drives degeneration and tau secretion in an in situ cellular tauopathy model. In “Animal Models of Alzheimer's Disease” Journal of Alzheimer’s Disease 2011. in press Hindawi ISBN: 978-953-307-797-0.
- Yamada K, Cirrito JR, Stewart FR, Jiang H, Finn MB, Holmes BB, et al. In vivo microdialysis reveals age-dependent decrease of brain interstitial fluid tau levels in P301S human tau transgenic mice. J Neurosci 2011; 31:13110 - 7; http://dx.doi.org/10.1523/JNEUROSCI.2569-11.2011; PMID: 21917794
- Frost B, Jacks RL, Diamond MI. Propagation of tau misfolding from the outside to the inside of a cell. J Biol Chem 2009; 284:12845 - 52; http://dx.doi.org/10.1074/jbc.M808759200; PMID: 19282288
- Hall GF, Lee S, Yao J. Neurofibrillary degeneration can be arrested in an in vivo cellular model of human tauopathy by application of a compound which inhibits tau filament formation in vitro. J Mol Neurosci 2002; 19:253 - 60; http://dx.doi.org/10.1385/JMN:19:3:251; PMID: 12540050
- Clavaguera F, Bolmont T, Crowther RA, Abramowski D, Frank S, Probst A, et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol 2009; 11:909 - 13; http://dx.doi.org/10.1038/ncb1901; PMID: 19503072
- Gómez-Ramos A, Díaz-Hernández M, Cuadros R, Hernández F, Avila J. Extracellular tau is toxic to neuronal cells. FEBS Lett 2006; 580:4842 - 50; http://dx.doi.org/10.1016/j.febslet.2006.07.078; PMID: 16914144
- Alper T, Cramp WA, Haig DA, Clarke MC. Does the agent of scrapie replicate without nucleic acid?. Nature 1967; 214:764 - 6; http://dx.doi.org/10.1038/214764a0; PMID: 4963878
- Gajdusek DC, Gibbs CJ, Alpers M. Experimental transmission of a Kuru-like syndrome to chimpanzees. Nature 1966; 209:794 - 6; http://dx.doi.org/10.1038/209794a0; PMID: 5922150
- Maddelein ML, Wickner RB. Two prion-inducing regions of Ure2p are nonoverlapping. Mol Cell Biol 1999; 19:4516 - 24; PMID: 10330190
- Masison DC, Wickner RB. Prion-inducing domain of yeast Ure2p and protease resistance of Ure2p in prion-containing cells. Science 1995; 270:93 - 5; http://dx.doi.org/10.1126/science.270.5233.93; PMID: 7569955
- Kabani M, Melki R. Yeast prions assembly and propagation: Contributions of the prion and non-prion moieties and the nature of assemblies. Prion 2011; 5; http://dx.doi.org/10.4161/pri.5.4.18070; PMID: 22052349
- Tuite MF, Serio TR. The prion hypothesis: from biological anomaly to basic regulatory mechanism. Nat Rev Mol Cell Biol 2010; 11:823 - 33; http://dx.doi.org/10.1038/nrm3007; PMID: 21081963
- Ross ED, Edskes HK, Terry MJ, Wickner RB. Primary sequence independence for prion formation. Proc Natl Acad Sci U S A 2005; 102:12825 - 30; http://dx.doi.org/10.1073/pnas.0506136102; PMID: 16123127
- Tanaka M, Chien P, Naber N, Cooke R, Weissman JS. Conformational variations in an infectious protein determine prion strain differences. Nature 2004; 428:323 - 8; http://dx.doi.org/10.1038/nature02392; PMID: 15029196
- Legname G, Nguyen HO, Baskakov IV, Cohen FE, Dearmond SJ, Prusiner SB. Strain-specified characteristics of mouse synthetic prions. Proc Natl Acad Sci U S A 2005; 102:2168 - 73; http://dx.doi.org/10.1073/pnas.0409079102; PMID: 15671162
- Nilsson KP, Joshi-Barr S, Winson O, Sigurdson CJ. Prion strain interactions are highly selective. J Neurosci 2010; 30:12094 - 102; http://dx.doi.org/10.1523/JNEUROSCI.2417-10.2010; PMID: 20826672
- Gambetti P, Cali I, Notari S, Kong Q, Zou WQ, Surewicz WK. Molecular biology and pathology of prion strains in sporadic human prion diseases. Acta Neuropathol 2011; 121:79 - 90; http://dx.doi.org/10.1007/s00401-010-0761-3; PMID: 21058033
- Zahn R. The octapeptide repeats in mammalian prion protein constitute a pH-dependent folding and aggregation site. J Mol Biol 2003; 334:477 - 88; http://dx.doi.org/10.1016/j.jmb.2003.09.048; PMID: 14623188
- Miller MB, Geoghegan JC, Supattapone S. Dissociation of infectivity from seeding ability in prions with alternate docking mechanism. PLoS Pathog 2011; 7:e1002128; http://dx.doi.org/10.1371/journal.ppat.1002128; PMID: 21779169
- Sandberg MK, Al-Doujaily H, Sharps B, Clarke AR, Collinge J. Prion propagation and toxicity in vivo occur in two distinct mechanistic phases. Nature 2011; 470:540 - 2; http://dx.doi.org/10.1038/nature09768; PMID: 21350487
- Linden R, Martins VR, Prado MA, Cammarota M, Izquierdo I, Brentani RR. Physiology of the prion protein. Physiol Rev 2008; 88:673 - 728; http://dx.doi.org/10.1152/physrev.00007.2007; PMID: 18391177
- Bodrikov V, Solis GP, Stuermer CA. Prion protein promotes growth cone development through reggie/flotillin-dependent N-cadherin trafficking. J Neurosci 2011; 31:18013 - 25; http://dx.doi.org/10.1523/JNEUROSCI.4729-11.2011; PMID: 22159115
- Klein C, Kramer EM, Cardine AM, Schraven B, Brandt R, Trotter J. Process outgrowth of oligodendrocytes is promoted by interaction of fyn kinase with the cytoskeletal protein tau. J Neurosci 2002; 22:698 - 707; PMID: 11826099
- Crozet C, Beranger F, Lehmann S. Cellular pathogenesis in prion diseases. Vet Res 2008; 39:44; http://dx.doi.org/10.1051/vetres:2008021; PMID: 18413130
- Bate C, Williams A. The cellular prion protein with a monoacylated glycosylphosphatidylinositol anchor modifies cell membranes, inhibits cell signaling and reduces prion formation. Prion 2011; 5:65 - 8; http://dx.doi.org/10.4161/pri.5.2.16095; PMID: 21738009
- Barria MA, Mukherjee A, Gonzalez-Romero D, Morales R, Soto C. De novo generation of infectious prions in vitro produces a new disease phenotype. PLoS Pathog 2009; 5:e1000421; http://dx.doi.org/10.1371/journal.ppat.1000421; PMID: 19436715
- Chesebro B, Race B, Meade-White K, Lacasse R, Race R, Klingeborn M, et al. Fatal transmissible amyloid encephalopathy: a new type of prion disease associated with lack of prion protein membrane anchoring. PLoS Pathog 2010; 6:e1000800; http://dx.doi.org/10.1371/journal.ppat.1000800; PMID: 20221436
- Chesebro B, Trifilo M, Race R, Meade-White K, Teng C, LaCasse R, et al. Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science 2005; 308:1435 - 9; http://dx.doi.org/10.1126/science.1110837; PMID: 15933194
- Sunyach C, Jen A, Deng J, Fitzgerald KT, Frobert Y, Grassi J, et al. The mechanism of internalization of glycosylphosphatidylinositol-anchored prion protein. EMBO J 2003; 22:3591 - 601; http://dx.doi.org/10.1093/emboj/cdg344; PMID: 12853474
- Mouillet-Richard S, Ermonval M, Chebassier C, Laplanche JL, Lehmann S, Launay JM, et al. Signal transduction through prion protein. Science 2000; 289:1925 - 8; http://dx.doi.org/10.1126/science.289.5486.1925; PMID: 10988071
- Si K, Giustetto M, Etkin A, Hsu R, Janisiewicz AM, Miniaci MC, et al. A neuronal isoform of CPEB regulates local protein synthesis and stabilizes synapse-specific long-term facilitation in aplysia. Cell 2003; 115:893 - 904; http://dx.doi.org/10.1016/S0092-8674(03)01021-3; PMID: 14697206
- Heinrich SU, Lindquist S. Protein-only mechanism induces self-perpetuating changes in the activity of neuronal Aplysia cytoplasmic polyadenylation element binding protein (CPEB). Proc Natl Acad Sci U S A 2011; 108:2999 - 3004; http://dx.doi.org/10.1073/pnas.1019368108; PMID: 21270333
- Si K, Lindquist S, Kandel ER. A neuronal isoform of the aplysia CPEB has prion-like properties. Cell 2003; 115:879 - 91; http://dx.doi.org/10.1016/S0092-8674(03)01020-1; PMID: 14697205
- Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L, et al. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci U S A 2009; 106:13010 - 5; http://dx.doi.org/10.1073/pnas.0903691106; PMID: 19651612
- Eisele YS, Obermüller U, Heilbronner G, Baumann F, Kaeser SA, Wolburg H, et al. Peripherally applied Abeta-containing inoculates induce cerebral beta-amyloidosis. Science 2010; 330:980 - 2; http://dx.doi.org/10.1126/science.1194516; PMID: 20966215
- Emmanouilidou E, Melachroinou K, Roumeliotis T, Garbis SD, Ntzouni M, Margaritis LH, et al. Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J Neurosci 2010; 30:6838 - 51; http://dx.doi.org/10.1523/JNEUROSCI.5699-09.2010; PMID: 20484626
- Rajendran L, Honsho M, Zahn TR, Keller P, Geiger KD, Verkade P, et al. Alzheimer’s disease beta-amyloid peptides are released in association with exosomes. Proc Natl Acad Sci U S A 2006; 103:11172 - 7; http://dx.doi.org/10.1073/pnas.0603838103; PMID: 16837572
- Lee HJ, Choi C, Lee SJ. Membrane-bound alpha-synuclein has a high aggregation propensity and the ability to seed the aggregation of the cytosolic form. J Biol Chem 2002; 277:671 - 8; http://dx.doi.org/10.1074/jbc.M107045200; PMID: 11679584
- Guo JL, Lee VM. Seeding of normal Tau by pathological Tau conformers drives pathogenesis of Alzheimer-like tangles. J Biol Chem 2011; 286:15317 - 31; http://dx.doi.org/10.1074/jbc.M110.209296; PMID: 21372138
- Corsetti V, Amadoro G, Gentile A, Capsoni S, Ciotti MT, Cencioni MT, et al. Identification of a caspase-derived N-terminal tau fragment in cellular and animal Alzheimer’s disease models. Mol Cell Neurosci 2008; 38:381 - 92; http://dx.doi.org/10.1016/j.mcn.2008.03.011; PMID: 18511295
- Binder LI, Frankfurter A, Rebhun LI. The distribution of tau in the mammalian central nervous system. J Cell Biol 1985; 101:1371 - 8; http://dx.doi.org/10.1083/jcb.101.4.1371; PMID: 3930508
- Lindwall G, Cole RD. Phosphorylation affects the ability of tau protein to promote microtubule assembly. J Biol Chem 1984; 259:5301 - 5; PMID: 6425287
- Drechsel DN, Hyman AA, Cobb MH, Kirschner MW. Modulation of the dynamic instability of tubulin assembly by the microtubule-associated protein tau. Mol Biol Cell 1992; 3:1141 - 54; PMID: 1421571
- Caceres A, Kosik KS. Inhibition of neurite polarity by tau antisense oligonucleotides in primary cerebellar neurons. Nature 1990; 343:461 - 3; http://dx.doi.org/10.1038/343461a0; PMID: 2105469
- Brandt R, Léger J, Lee G. Interaction of tau with the neural plasma membrane mediated by tau’s amino-terminal projection domain. J Cell Biol 1995; 131:1327 - 40; http://dx.doi.org/10.1083/jcb.131.5.1327; PMID: 8522593
- Lee G, Newman ST, Gard DL, Band H, Panchamoorthy G. Tau interacts with src-family non-receptor tyrosine kinases. J Cell Sci 1998; 111:3167 - 77; PMID: 9763511
- Belkadi A, LoPresti P. Truncated Tau with the Fyn-binding domain and without the microtubule-binding domain hinders the myelinating capacity of an oligodendrocyte cell line. J Neurochem 2008; 107:351 - 60; http://dx.doi.org/10.1111/j.1471-4159.2008.05600.x; PMID: 18680553
- Zmuda JF, Rivas RJ. Actin disruption alters the localization of tau in the growth cones of cerebellar granule neurons. J Cell Sci 2000; 113:2797 - 809; PMID: 10893194
- Sverdlov M, Shajahan AN, Minshall RD. Tyrosine phosphorylation-dependence of caveolae-mediated endocytosis. J Cell Mol Med 2007; 11:1239 - 50; http://dx.doi.org/10.1111/j.1582-4934.2007.00127.x; PMID: 18205698
- Hall GF, Yao J, Lee G. Human tau becomes phosphorylated and forms filamentous deposits when overexpressed in lamprey central neurons in situ. Proc Natl Acad Sci U S A 1997; 94:4733 - 8; http://dx.doi.org/10.1073/pnas.94.9.4733; PMID: 9114060
- Hall GF, Chu B, Lee G, Yao J. Human tau filaments induce microtubule and synapse loss in an in vivo model of neurofibrillary degenerative disease. J Cell Sci 2000; 113:1373 - 87; PMID: 10725221
- Hall GF, Lee VM, Lee G, Yao J. Staging of neurofibrillary degeneration caused by human tau overexpression in a unique cellular model of human tauopathy. Am J Pathol 2001; 158:235 - 46; http://dx.doi.org/10.1016/S0002-9440(10)63962-4; PMID: 11141497
- Honson NS, Jensen JR, Abraha A, Hall GF, Kuret J. Small-molecule mediated neuroprotection in an in situ model of tauopathy. Neurotox Res 2009; 15:274 - 83; http://dx.doi.org/10.1007/s12640-009-9028-y; PMID: 19384600
- Gómez-Ramos A, Díaz-Hernández M, Rubio A, Miras-Portugal MT, Avila J. Extracellular tau promotes intracellular calcium increase through M1 and M3 muscarinic receptors in neuronal cells. Mol Cell Neurosci 2008; 37:673 - 81; http://dx.doi.org/10.1016/j.mcn.2007.12.010; PMID: 18272392
- Iliev AI, Ganesan S, Bunt G, Wouters FS. Removal of pattern-breaking sequences in microtubule binding repeats produces instantaneous tau aggregation and toxicity. J Biol Chem 2006; 281:37195 - 204; http://dx.doi.org/10.1074/jbc.M604863200; PMID: 17008320
- Mocanu MM, Nissen A, Eckermann K, Khlistunova I, Biernat J, Drexler D, et al. The potential for beta-structure in the repeat domain of tau protein determines aggregation, synaptic decay, neuronal loss, and coassembly with endogenous Tau in inducible mouse models of tauopathy. J Neurosci 2008; 28:737 - 48; http://dx.doi.org/10.1523/JNEUROSCI.2824-07.2008; PMID: 18199773
- Arima K. Ultrastructural characteristics of tau filaments in tauopathies: immuno-electron microscopic demonstration of tau filaments in tauopathies. Neuropathology 2006; 26:475 - 83; http://dx.doi.org/10.1111/j.1440-1789.2006.00669.x; PMID: 17080728
- van Eersel J, Bi M, Ke YD, Hodges JR, Xuereb JH, Gregory GC, et al. Phosphorylation of soluble tau differs in Pick’s disease and Alzheimer’s disease brains. J Neural Transm 2009; 116:1243 - 51; http://dx.doi.org/10.1007/s00702-009-0293-y; PMID: 19693433
- Sergeant N, Wattez A, Delacourte A. Neurofibrillary degeneration in progressive supranuclear palsy and corticobasal degeneration: tau pathologies with exclusively “exon 10” isoforms. J Neurochem 1999; 72:1243 - 9; http://dx.doi.org/10.1046/j.1471-4159.1999.0721243.x; PMID: 10037497
- Chirita CN, Necula M, Kuret J. Anionic micelles and vesicles induce tau fibrillization in vitro. J Biol Chem 2003; 278:25644 - 50; http://dx.doi.org/10.1074/jbc.M301663200; PMID: 12730214
- Díaz-Nido J, Wandosell F, Avila J. Glycosaminoglycans and beta-amyloid, prion and tau peptides in neurodegenerative diseases. Peptides 2002; 23:1323 - 32; http://dx.doi.org/10.1016/S0196-9781(02)00068-2; PMID: 12128089
- Alonso AD, Grundke-Iqbal I, Barra HS, Iqbal K. Abnormal phosphorylation of tau and the mechanism of Alzheimer neurofibrillary degeneration: sequestration of microtubule-associated proteins 1 and 2 and the disassembly of microtubules by the abnormal tau. Proc Natl Acad Sci U S A 1997; 94:298 - 303; http://dx.doi.org/10.1073/pnas.94.1.298; PMID: 8990203
- Park SY, Ferreira A. The generation of a 17 kDa neurotoxic fragment: an alternative mechanism by which tau mediates beta-amyloid-induced neurodegeneration. J Neurosci 2005; 25:5365 - 75; http://dx.doi.org/10.1523/JNEUROSCI.1125-05.2005; PMID: 15930385
- Amadoro G, Ciotti MT, Costanzi M, Cestari V, Calissano P, Canu N. NMDA receptor mediates tau-induced neurotoxicity by calpain and ERK/MAPK activation. Proc Natl Acad Sci U S A 2006; 103:2892 - 7; http://dx.doi.org/10.1073/pnas.0511065103; PMID: 16477009
- King ME, Kan HM, Baas PW, Erisir A, Glabe CG, Bloom GS. Tau-dependent microtubule disassembly initiated by prefibrillar beta-amyloid. J Cell Biol 2006; 175:541 - 6; http://dx.doi.org/10.1083/jcb.200605187; PMID: 17101697
- Rapoport M, Dawson HN, Binder LI, Vitek MP, Ferreira A. Tau is essential to beta -amyloid-induced neurotoxicity. Proc Natl Acad Sci U S A 2002; 99:6364 - 9; http://dx.doi.org/10.1073/pnas.092136199; PMID: 11959919
- Wang YP, Biernat J, Pickhardt M, Mandelkow E, Mandelkow EM. Stepwise proteolysis liberates tau fragments that nucleate the Alzheimer-like aggregation of full-length tau in a neuronal cell model. Proc Natl Acad Sci U S A 2007; 104:10252 - 7; http://dx.doi.org/10.1073/pnas.0703676104; PMID: 17535890
- Zilka N, Filipcik P, Koson P, Fialova L, Skrabana R, Zilkova M, et al. Truncated tau from sporadic Alzheimer’s disease suffices to drive neurofibrillary degeneration in vivo. FEBS Lett 2006; 580:3582 - 8; http://dx.doi.org/10.1016/j.febslet.2006.05.029; PMID: 16753151
- Baum L, Seger R, Woodgett JR, Kawabata S, Maruyama K, Koyama M, et al. Overexpressed tau protein in cultured cells is phosphorylated without formation of PHF: implication of phosphoprotein phosphatase involvement. Brain Res Mol Brain Res 1995; 34:1 - 17; http://dx.doi.org/10.1016/0169-328X(95)00111-5; PMID: 8750856
- Merrick SE, Demoise DC, Lee VM. Site-specific dephosphorylation of tau protein at Ser202/Thr205 in response to microtubule depolymerization in cultured human neurons involves protein phosphatase 2A. J Biol Chem 1996; 271:5589 - 94; http://dx.doi.org/10.1074/jbc.271.10.5589; PMID: 8621419
- Ishihara T, Hong M, Zhang B, Nakagawa Y, Lee MK, Trojanowski JQ, et al. Age-dependent emergence and progression of a tauopathy in transgenic mice overexpressing the shortest human tau isoform. Neuron 1999; 24:751 - 62; http://dx.doi.org/10.1016/S0896-6273(00)81127-7; PMID: 10595524
- Spittaels K, Van den Haute C, Van Dorpe J, Bruynseels K, Vandezande K, Laenen I, et al. Prominent axonopathy in the brain and spinal cord of transgenic mice overexpressing four-repeat human tau protein. Am J Pathol 1999; 155:2153 - 65; http://dx.doi.org/10.1016/S0002-9440(10)65533-2; PMID: 10595944
- Hall GF, Yao J. Modeling tauopathy: a range of complementary approaches. Biochim Biophys Acta 2005; 1739:224 - 39; PMID: 15615641
- Simón D, García-García E, Royo F, Falcón-Pérez JM, Avila J. Proteostasis of tau. Tau overexpression results in its secretion via membrane vesicles. FEBS Lett 2012; 586:47 - 54; http://dx.doi.org/10.1016/j.febslet.2011.11.022; PMID: 22138183
- Götz J, Probst A, Spillantini MG, Schäfer T, Jakes R, Bürki K, et al. Somatodendritic localization and hyperphosphorylation of tau protein in transgenic mice expressing the longest human brain tau isoform. EMBO J 1995; 14:1304 - 13; PMID: 7729409
- Brion JP, Tremp G, Octave JN. Transgenic expression of the shortest human tau affects its compartmentalization and its phosphorylation as in the pretangle stage of Alzheimer’s disease. Am J Pathol 1999; 154:255 - 70; http://dx.doi.org/10.1016/S0002-9440(10)65272-8; PMID: 9916940
- Lewis J, McGowan E, Rockwood J, Melrose H, Nacharaju P, Van Slegtenhorst M, et al. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet 2000; 25:402 - 5; http://dx.doi.org/10.1038/78078; PMID: 10932182
- Busciglio J, Lorenzo A, Yeh J, Yankner BA. beta-amyloid fibrils induce tau phosphorylation and loss of microtubule binding. Neuron 1995; 14:879 - 88; http://dx.doi.org/10.1016/0896-6273(95)90232-5; PMID: 7718249
- Hong M, Zhukareva V, Vogelsberg-Ragaglia V, Wszolek Z, Reed L, Miller BI, et al. Mutation-specific functional impairments in distinct tau isoforms of hereditary FTDP-17. Science 1998; 282:1914 - 7; http://dx.doi.org/10.1126/science.282.5395.1914; PMID: 9836646
- Nacharaju P, Lewis J, Easson C, Yen S, Hackett J, Hutton M, et al. Accelerated filament formation from tau protein with specific FTDP-17 missense mutations. FEBS Lett 1999; 447:195 - 9; http://dx.doi.org/10.1016/S0014-5793(99)00294-X; PMID: 10214944
- Nickel W. Unconventional secretory routes: direct protein export across the plasma membrane of mammalian cells. Traffic 2005; 6:607 - 14; http://dx.doi.org/10.1111/j.1600-0854.2005.00302.x; PMID: 15998317
- van Harten AC, Kester MI, Visser PJ, Blankenstein MA, Pijnenburg YA, van der Flier WM, et al. Tau and p-tau as CSF biomarkers in dementia: a meta-analysis. Clin Chem Lab Med 2011; 49:353 - 66; http://dx.doi.org/10.1515/cclm.2011.086; PMID: 21342021
- Johnson GV, Seubert P, Cox TM, Motter R, Brown JP, Galasko D. The tau protein in human cerebrospinal fluid in Alzheimer’s disease consists of proteolytically derived fragments. J Neurochem 1997; 68:430 - 3; http://dx.doi.org/10.1046/j.1471-4159.1997.68010430.x; PMID: 8978756
- Schiess MC, Barnes JL, Ellmore TM, Poindexter BJ, Dinh K, Bick RJ. CSF from Parkinson disease patients differentially affects cultured microglia and astrocytes. BMC Neurosci 2010; 11:151; PMID: 21114836
- Diaz-Espinoza R, Soto C. Generation of prions in vitro and the protein-only hypothesis. Prion 2010; 4:53 - 9; http://dx.doi.org/10.4161/pri.4.2.11960; PMID: 20448454
- Ryou C, Mays CE. Prion propagation in vitro: are we there yet?. Int J Med Sci 2008; 5:347 - 53; http://dx.doi.org/10.7150/ijms.5.347; PMID: 19015743
- Graham JF, Agarwal S, Kurian D, Kirby L, Pinheiro TJ, Gill AC. Low density subcellular fractions enhance disease-specific prion protein misfolding. J Biol Chem 2010; 285:9868 - 80; http://dx.doi.org/10.1074/jbc.M109.093484; PMID: 20106973
- Wang XF, Dong CF, Zhang J, Wan YZ, Li F, Huang YX, et al. Human tau protein forms complex with PrP and some GSS- and fCJD-related PrP mutants possess stronger binding activities with tau in vitro. Mol Cell Biochem 2008; 310:49 - 55; http://dx.doi.org/10.1007/s11010-007-9664-6; PMID: 18038270
- Lawson VA, Klemm HM, Welton JM, Masters CL, Crouch P, Cappai R, et al. Gene knockout of tau expression does not contribute to the pathogenesis of prion disease. J Neuropathol Exp Neurol 2011; 70:1036 - 45; http://dx.doi.org/10.1097/NEN.0b013e318235b471; PMID: 22002429
- Canu N, Filesi I, Pristerà A, Ciotti MT, Biocca S. Altered intracellular distribution of PrP(C) and impairment of proteasome activity in tau overexpressing cortical neurons. J Alzheimers Dis 2011; 27:603 - 13; PMID: 21841253
- Lee S, Jung C, Lee G, Hall GF. Exonic point mutations of human tau enhance its toxicity and cause characteristic changes in neuronal morphology, tau distribution and tau phosphorylation in the lamprey cellular model of tauopathy. J Alzheimers Dis 2009; 16:99 - 111; PMID: 19158426
- Caughey B, Baron GS, Chesebro B, Jeffrey M. Getting a grip on prions: oligomers, amyloids, and pathological membrane interactions. Annu Rev Biochem 2009; 78:177 - 204; http://dx.doi.org/10.1146/annurev.biochem.78.082907.145410; PMID: 19231987
- Santuccione A, Sytnyk V, Leshchyns’ka I, Schachner M. Prion protein recruits its neuronal receptor NCAM to lipid rafts to activate p59fyn and to enhance neurite outgrowth. J Cell Biol 2005; 169:341 - 54; http://dx.doi.org/10.1083/jcb.200409127; PMID: 15851519
- Bizat N, Peyrin JM, Haïk S, Cochois V, Beaudry P, Laplanche JL, et al. Neuron dysfunction is induced by prion protein with an insertional mutation via a Fyn kinase and reversed by sirtuin activation in Caenorhabditis elegans. J Neurosci 2010; 30:5394 - 403; http://dx.doi.org/10.1523/JNEUROSCI.5831-09.2010; PMID: 20392961
- Fevrier B, Vilette D, Archer F, Loew D, Faigle W, Vidal M, et al. Cells release prions in association with exosomes. Proc Natl Acad Sci U S A 2004; 101:9683 - 8; http://dx.doi.org/10.1073/pnas.0308413101; PMID: 15210972
- Vella LJ, Sharples RA, Lawson VA, Masters CL, Cappai R, Hill AF. Packaging of prions into exosomes is associated with a novel pathway of PrP processing. J Pathol 2007; 211:582 - 90; http://dx.doi.org/10.1002/path.2145; PMID: 17334982
- Fang Y, Wu N, Gan X, Yan W, Morrell JC, Gould SJ. Higher-order oligomerization targets plasma membrane proteins and HIV gag to exosomes. PLoS Biol 2007; 5:e158; http://dx.doi.org/10.1371/journal.pbio.0050158; PMID: 17550307
- Nunziante M, Ackermann K, Dietrich K, Wolf H, Gädtke L, Gilch S, et al. Proteasomal dysfunction and endoplasmic reticulum stress enhance trafficking of prion protein aggregates through the secretory pathway and increase accumulation of pathologic prion protein. J Biol Chem 2011; 286:33942 - 53; http://dx.doi.org/10.1074/jbc.M111.272617; PMID: 21835918
- Gadad BS, Britton GB, Rao KS. Targeting oligomers in neurodegenerative disorders: lessons from α-synuclein, tau, and amyloid-β peptide. J Alzheimers Dis 2011; 24:Suppl 2 223 - 32; PMID: 21460436
- Deleault NR, Lucassen RW, Supattapone S. RNA molecules stimulate prion protein conversion. Nature 2003; 425:717 - 20; http://dx.doi.org/10.1038/nature01979; PMID: 14562104
- Kampers T, Friedhoff P, Biernat J, Mandelkow EM, Mandelkow E. RNA stimulates aggregation of microtubule-associated protein tau into Alzheimer-like paired helical filaments. FEBS Lett 1996; 399:344 - 9; http://dx.doi.org/10.1016/S0014-5793(96)01386-5; PMID: 8985176
- Zehe C, Engling A, Wegehingel S, Schäfer T, Nickel W. Cell-surface heparan sulfate proteoglycans are essential components of the unconventional export machinery of FGF-2. Proc Natl Acad Sci U S A 2006; 103:15479 - 84; http://dx.doi.org/10.1073/pnas.0605997103; PMID: 17030799
- Peters PJ, Mironov A Jr., Peretz D, van Donselaar E, Leclerc E, Erpel S, et al. Trafficking of prion proteins through a caveolae-mediated endosomal pathway. J Cell Biol 2003; 162:703 - 17; http://dx.doi.org/10.1083/jcb.200304140; PMID: 12925711
- Kremer A, Maurin H, Demedts D, Devijver H, Borghgraef P, Van Leuven F. Early improved and late defective cognition is reflected by dendritic spines in Tau.P301L mice. J Neurosci 2011; 31:18036 - 47; http://dx.doi.org/10.1523/JNEUROSCI.4859-11.2011; PMID: 22159117
- Boekhoorn K, Terwel D, Biemans B, Borghgraef P, Wiegert O, Ramakers GJ, et al. Improved long-term potentiation and memory in young tau-P301L transgenic mice before onset of hyperphosphorylation and tauopathy. J Neurosci 2006; 26:3514 - 23; http://dx.doi.org/10.1523/JNEUROSCI.5425-05.2006; PMID: 16571759
- Tackenberg C, Brandt R. Divergent pathways mediate spine alterations and cell death induced by amyloid-beta, wild-type tau, and R406W tau. J Neurosci 2009; 29:14439 - 50; http://dx.doi.org/10.1523/JNEUROSCI.3590-09.2009; PMID: 19923278
- Perrin RJ, Woods WS, Clayton DF, George JM. Exposure to long chain polyunsaturated fatty acids triggers rapid multimerization of synucleins. J Biol Chem 2001; 276:41958 - 62; http://dx.doi.org/10.1074/jbc.M105022200; PMID: 11553616
- Sluchanko NN, Gusev NB. Probable participation of 14-3-3 in tau protein oligomerization and aggregation. J Alzheimers Dis 2011; 27:467 - 76; PMID: 21876254
- Waxman EA, Giasson BI. Induction of intracellular tau aggregation is promoted by α-synuclein seeds and provides novel insights into the hyperphosphorylation of tau. J Neurosci 2011; 31:7604 - 18; http://dx.doi.org/10.1523/JNEUROSCI.0297-11.2011; PMID: 21613474
- Whitwell JL, Jack CR Jr., Parisi JE, Knopman DS, Boeve BF, Petersen RC, et al. Rates of cerebral atrophy differ in different degenerative pathologies. Brain 2007; 130:1148 - 58; http://dx.doi.org/10.1093/brain/awm021; PMID: 17347250
- Esposito A, Dohm CP, Kermer P, Bähr M, Wouters FS. alpha-Synuclein and its disease-related mutants interact differentially with the microtubule protein tau and associate with the actin cytoskeleton. Neurobiol Dis 2007; 26:521 - 31; http://dx.doi.org/10.1016/j.nbd.2007.01.014; PMID: 17408955
- Chen C, Shi Q, Zhang BY, Wang GR, Zhou W, Gao C, et al. The prepared tau exon-specific antibodies revealed distinct profiles of tau in CSF of the patients with Creutzfeldt-Jakob disease. PLoS One 2010; 5:e11886; http://dx.doi.org/10.1371/journal.pone.0011886; PMID: 20686702
- Lasagna-Reeves CA, Castillo-Carranza DL, Guerrero-Muoz MJ, Jackson GR, Kayed R. Preparation and characterization of neurotoxic tau oligomers. Biochemistry 2010; 49:10039 - 41; http://dx.doi.org/10.1021/bi1016233; PMID: 21047142
- Terwel D, Lasrado R, Snauwaert J, Vandeweert E, Van Haesendonck C, Borghgraef P, et al. Changed conformation of mutant Tau-P301L underlies the moribund tauopathy, absent in progressive, nonlethal axonopathy of Tau-4R/2N transgenic mice. J Biol Chem 2005; 280:3963 - 73; http://dx.doi.org/10.1074/jbc.M409876200; PMID: 15509565
- Liu L, Drouet V, Wu JW, Witter MP, Small SA, Clelland C, et al. Trans-synaptic spread of tau pathology in vivo. PLoS One 2012; 7:e31302; http://dx.doi.org/10.1371/journal.pone.0031302; PMID: 22312444
- Alais S, Simoes S, Baas D, Lehmann S, Raposo G, Darlix JL, et al. Mouse neuroblastoma cells release prion infectivity associated with exosomal vesicles. Biol Cell 2008; 100:603 - 15; http://dx.doi.org/10.1042/BC20080025; PMID: 18422484
- Veith NM, Plattner H, Stuermer CA, Schulz-Schaeffer WJ, Bürkle A. Immunolocalisation of PrPSc in scrapie-infected N2a mouse neuroblastoma cells by light and electron microscopy. Eur J Cell Biol 2009; 88:45 - 63; http://dx.doi.org/10.1016/j.ejcb.2008.08.001; PMID: 18834644
- Estes PS, Boehringer A, Zwick R, Tang JE, Grigsby B, Zarnescu DC. Wild-type and A315T mutant TDP-43 exert differential neurotoxicity in a Drosophila model of ALS. Hum Mol Genet 2011; 20:2308 - 21; http://dx.doi.org/10.1093/hmg/ddr124; PMID: 21441568