
Abstract
Nucleic acid sequences of the prion gene (PRNP) were examined and genotypes compiled for 76 white-tailed deer from northern Illinois, which previously tested positive for chronic wasting disease (CWD), and 120 negative animals selected to control for geographic location and age. Nine nucleotide polymorphisms, seven silent and two coding, were found in the sampled population. All observed polymorphisms except two of very low frequency were observed in both negative and positive animals, although five polymorphic loci had significantly different distributions of alleles between infected and non-infected individuals. Nucleotide base changes 60C/T, 285A/C, 286G/A, and 555C/T were observed with higher than expected frequencies in CWD negative animals suggesting disease resistance, while 153C/T was observed more than expected in positive animals, suggesting susceptibility. The two coding polymorphisms, 285A/C (Q95H) and 286G/A (G96S), have been described in white-tailed deer populations sampled in Colorado and Wisconsin. Frequency distributions of coding polymorphisms in Wisconsin and Illinois deer populations were different, an unexpected result considering the sampled areas are less than 150 km apart. The total number of polymorphisms per animal, silent or coding, was negatively correlated to disease status. The potential importance of silent polymorphisms (60C/T, 153C/T, 555C/T), either individually or cumulatively, in CWD disease status has not been previously reported.
Acknowledgements
The Illinois Department of Natural Resources was responsible for the collection of all samples and this project could not have been completed without the numerous Wildlife Biologists who generously donated both their time and labor to this study. The Illinois Department of Agriculture Animal Disease Laboratory in Galesburg also provided invaluable assistance. Funding for this project was provided by the Illinois Department of Natural Resources, the United States Geological Survey, the Federal Aid in Wildlife Restoration Project W-146-R, the Jonathan Baldwin Turner Scholarship Fund and the Environmental Council SURE grant program.
Figures and Tables
Figure 1 Nucleotide and amino acid database consensus sequence and polymorphisms observed in Illinois white-tailed deer. Numbers indicate the nucleotide or deduced amino acid sequence from a consensus. Observed frequency (# of polymorphic alleles/total # of alleles* 100) of polymorphisms and domains within the prion protein are also indicated.
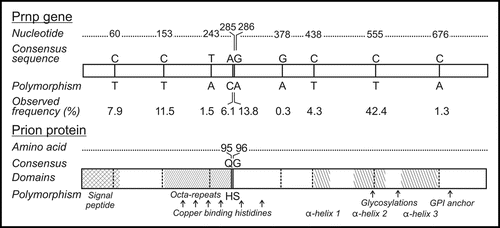
Figure 2 Probability of CWD as predicted by the number of polymorphic alleles. Total polymorphisms, silent polymorphisms, coding polymorphisms and total polymorphisms omitting locus 153 were used as independent variables in separate logistic regression models with CWD as the outcome variable. Odds ratios were estimated using the number of heterozygous and homozygous SNP summed across all loci. Coding for locus 153 was reversed because wild-type alleles conferred susceptibility (2 = homozygous for the consensus genotype, 1 = heterozygous, 0 = homozygous for the polymorphism), which resulted in a higher predicted probability of CWD than in a model omitting locus 153.
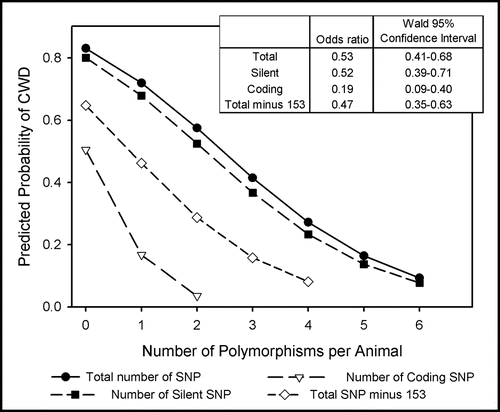
Figure 3 Distribution of coding polymorphisms Q95H and G96S. Bars represent the percent of sampled population of the indicated genotype within disease status. Distribution of coding polymorphisms for sampled populations of Illinois deer are represented by open bars. Distribution of polymorphisms for Wisconsin deer (hatched bars) were calculated from published results.Citation12
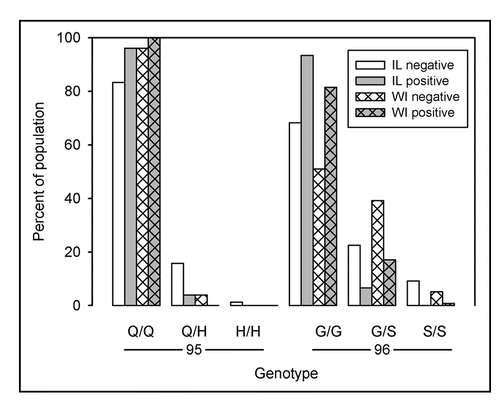
Table 1 Ratio of observed and expected allele frequencies for nucleotide polymorphisms
Table 2 Ratio of observed and expected genotype frequencies for nucleotide polymorphisms
Table 3 Reconstructed frequencies of common haplotypes for CWD positive animals and controls