Abstract
Prion protein (PrP)-like molecule, doppel (Dpl), is neurotoxic in mice, causing Purkinje cell degeneration. In contrast, PrP antagonizes Dpl in trans, rescuing mice from Purkinje cell death. We have previously shown that PrP with deletion of the N-terminal residues 23-88 failed to neutralize Dpl in mice, indicating that the N-terminal region, particularly that including residues 23-88, may have trans-protective activity against Dpl. Interestingly, PrP with deletion elongated to residues 121 or 134 in the N-terminal region was shown to be similarly neurotoxic to Dpl, indicating that the PrP C-terminal region may have toxicity which is normally prevented by the N-terminal domain in cis. We recently investigated further roles for the N-terminal region of PrP in antagonistic interactions with Dpl by producing three different types of transgenic mice. These mice expressed PrP with deletion of residues 25-50 or 51-90, or a fusion protein of the N-terminal region of PrP with Dpl. Here, we discuss a possible model for the antagonistic interaction between PrP and Dpl .
The normal prion protein, termed PrPC, is a membrane glycoprotein tethered to the outer cell surface via a glycosylphosphatidylinositol (GPI) anchor moiety.Citation1,Citation2 It is ubiquitously expressed in neuronal and non-neuronal tissues, with highest expression in the central nervous system, particularly in neurons.Citation3 The physiological function of PrPC remains elusive. We and others have shown that PrPC functionally antagonizes doppel (Dpl), a PrP-like GPI-anchored protein with ∼23% identity in amino acid composition to PrP, protecting Dpl-induced neurotoxicity in mice.Citation4–Citation7 Dpl is encoded on Prnd located downstream of the PrP gene (Prnp) and expressed in the testis, heart, kidney and spleen of wild-type mice but not in the brain where PrPC is actively expressed.Citation4,Citation5,Citation8 However, when ectopically expressed in brains, particularly in cerebellar Purkinje cells, Dpl exerts a neurotoxic activity, causing ataxia and Purkinje cell degeneration in Ngsk, Rcm0 and Zrch II lines of mice devoid of PrPC (Prnp0/0).Citation4,Citation9,Citation10 In these mice, Dpl was abnormally controlled by the upstream Prnp promoter.Citation4,Citation5 This is due to targeted deletion of part of Prnp including a splicing acceptor of exon 3.Citation11 Pre-mRNA starting from the residual exon1/2 of Prnp was abnormally elongated until the end of Prnd and then intergenically spliced between the residual Prnp exons 1/2 and the Prnd coding exons.Citation4,Citation5 As a result, Dpl was ectopically expressed under the control of the Prnp promoter in the brain, particularly in neurons including Purkinje cells.Citation4,Citation5 In contrast, in other Prnp0/0 lines, such as Zrch I and Npu, the splicing acceptor was intact, resulting in normal Purkinje cells without ectopic expression of Dpl in the brain.Citation4
The molecular mechanism of the antagonistic interaction between PrPC and Dpl remains unknown. We recently showed that the N-terminal half of PrPC includes elements that might mediate cis or trans protection against Dpl in mice, ameliorating Purkinje cell degeneration.Citation12 We also showed that the octapeptide repeat (OR) region in the N-terminal domain is dispensable for PrPC to neutralize Dpl neurotoxicity in mice.Citation12 Here, possible molecular mechanisms for the antagonism between PrPC and Dpl will be discussed.
Lack of a cis-Protective Element Renders PrP and Dpl Neurotoxic
PrPC largely comprises of two domains, the N-terminal and C-terminal domains (). The N-terminal domain is highly flexible, lacking identifiable secondary structures.Citation13 This domain includes the OR region, which is unique to all PrP molecules.Citation2 In contrast, the C-terminal domain forms a globular structure with three α-strands and two short β-strands.Citation13 Interestingly, PrP with the N-terminal residues 32–121 or 32–134 deleted, termed PrPΔ32–121 and PrPΔ32–134 (Constructs 1 and 2 in ), respectively, was shown to be neurotoxic.Citation14 This caused ataxia and cerebellar neurodegeneration, including granule or Purkinje cell death in Zrch I Prnp0/0 mice.Citation14,Citation15 These results suggest that PrPC is potentially neurotoxic via the C-terminal domain but under normal conditions the neurotoxicity of the C-terminal domain may be masked by the N-terminal domain.
Dpl is a homologue of the C-terminal globular domain of PrPC ().Citation16 However, Dpl lacks the amino acid sequences corresponding to the N-terminal half of PrPC (). It is therefore conceivable that the neurotoxicity of Dpl might be due to lack of the corresponding N-terminal part of PrPC. Consistent with this, we recently showed that PrPN-Dpl (Construct 3 in ), a fusion protein of the N-terminal residues 1–124 of PrPC and the residues 58–179 of Dpl, was itself non-toxic in mice.Citation12 It induced neither ataxia nor Purkinje cell degeneration in Zrch I Prnp0/0 mice, even when transgenically expressed in the brain under the control of the Prnp promoter.Citation12 Constructs 1 and 2 cover most of the Dpl-homologous C-terminal part of PrP. These observations strongly suggest that Dpl might undergo the same or very similar molecular processes as toxic PrP molecules do to perform their neurotoxicity in mice.
In contrast to the Constructs 1 and 2, PrPΔ23–88 (Construct 4 in ) is non-toxic in mice.Citation17 This indicates that the central region including residues 89–121, which are deleted in the toxic Constructs 1 and 2 but intact in the non-toxic Construct 4, may include an element(s) that mediates the cis-protection against the neurotoxic C-terminal domain. Indeed, PrP with deletion of the central resides 105–125 or 94–134 (Constructs 5 and 6 in ) was shown to be neurotoxic, causing cerebellar degeneration or demyelination in mice.Citation18,Citation19 However, no neurotoxicity was detected for PrP with deletion of only eight amino acids (residues 114–121) in the central region (Construct 7 in ).Citation18 These results suggest that the cis-protective activity of the central region might be regulated in a highly integrated way, which might be impaired by deletion of a large part of the region rather than any specific amino acids or small areas.
Trans-Protection by PrP Against Dpl
Trans-protective activity of various PrP constructs against Dpl or the toxic truncated PrPs is summarized in . Wild-type PrPC has the potential to abrogate Dpl neurotoxicity in trans. The ataxia and Purkinje cell degeneration, which were induced by transgenic expression of Dpl in the brain, could be attenuated in mice carrying the wild-type but not the knockout genetic background for Prnp.Citation6,Citation7 We previously showed that Construct 4, which lacks the N-terminal residues 23–88, completely lost the ability to rescue an ataxic Ngsk line of Prnp0/0 mice from Dpl-induced Purkinje cell degeneration.Citation17 We also recently showed that Construct 3 in which the PrP N terminal region (residues 23–124) was fused to Dpl (residues 58–179) mitigated the neurotoxicity of transgenically expressed wild-type Dpl in mice, prolonging the times to the onset of ataxia and Purkinje cell degeneration.Citation12 These results indicate that the N-terminal domain, particularly that encompassing residues 23–88, might include an element(s) that mediates the antagonistic function of PrPC against Dpl in trans. However, the trans-protective element might require cis-protective activity to function, because the neurotoxic Constructs 5 and 6 include the trans-elements but not the cis-element.Citation18,Citation19
Residues 23–88 cover the entire pre-OR and almost the entire OR except for two amino acids (residues 89 and 90). We recently investigated the role of the OR and the pre-OR in the trans-neuroprotection of PrPC against Dpl by producing transgenic mice expressing Constructs 8 or 9.Citation12 They expressed PrP with deletion of the entire OR (residues 51–90) or most of the pre-OR (residues 25–50) except for residues 23 and 24.Citation12 Complete rescue from ataxia and Purkinje cell degeneration was detected in mice co-expressing the OR-lacking Construct 8 and Dpl in the absence of wild-type PrPC,Citation12 clearly indicating that the OR is dispensable for PrPC protection against Dpl-neurotoxicity in trans. The pre-OR-lacking Construct 9 also blocked Dpl-neurotoxicity in mice in a manner dependent on its expression level, prolonging the onset of ataxia and Purkinje cell death.Citation12 Shmerling et al. reported that the cerebellar granule cell death induced by the neurotoxic Construct 2 in Zrch I Prnp0/0 mice could be abrogated by Construct 10.Citation14 Construct 10 lacks the entire OR and part of the pre-OR. These findings indicate that the OR and part of the pre-OR are also unnecessary for PrPC to antagonize the neurotoxicity of truncated PrPs in trans.
Two amino acids (residues 23 and 24) of the pre-OR are commonly intact in the trans-protective molecules, including wild-type PrPC and Constructs 3 as well as 8–10, but not in the non-protective Construct 4. It is therefore possible that these two residues are important for the trans-neuroprotection of PrPC against Dpl or the toxic truncated PrPs. Interestingly, the two amino acids are followed by residues starting from 51 in Construct 9, generating a new N-terminal sequence (KKPQGGTWG), which is very similar to the N-terminal 9 residues (KKRPKPGGW) of wild-type PrPC. Six out of nine of these amino acids are identical. It is thus possible that this newly generated N-terminal sequence might mimic the function of wild-type N-terminal 9 residues. This N-terminal sequence also remains intact in the other protective Constructs 3, 8 and 10. This therefore suggests that rather than the two amino acids, the 9 N-terminal residues may be relevant to the trans-neuroprotection of PrPC against Dpl. It might be alternatively possible that the trans-neuroprotection of PrPC against Dpl may be impaired only by a large deletion of the N-terminal domain, such as deletion of the residues 23–88, but not by small deletions such as deletion of part of pre-OR and/or OR.
Possible Mechanism of Antagonistic Interaction between PrP and Dpl
The exact mechanism by which PrPC antagonizes Dpl, preventing Purkinje cell degeneration, remains elusive. Accumulating evidence indicates that PrPC might function as a neuroprotective molecule by exerting anti-apoptotic activities. Indeed, we and others showed that Prnp0/0 mice were highly sensitive to ischemic or traumatic brain damage, developing more severe apoptotic neuronal cell death than in wild-type mice.Citation20–Citation23 Moreover, it was reported that hippocampal neuronal cell lines established from Prnp0/0 mice easily succumbed to apoptosis after serum withdrawal, and that expression of either PrPC or the anti-apoptotic molecule Bcl-2 rescued cell lines from the apoptosis.Citation24 PrPC also prevented Bax-induced apoptosis in human primary neurons.Citation25 Interestingly, PrP lacking OR failed to rescue the cells from the apoptosis, which was induced by serum withdrawal or Bax, and Zrch I Prnp0/0 mice from ischemic brain damage.Citation25–Citation27 This indicates that the OR is essential for the neuroprotective activity of PrPC. However, we demonstrated that the OR is dispensable for PrPC to antagonize Dpl in mice.Citation12 Shmerling et al. also showed that the OR is unnecessary for PrPC to antagonize the neurotoxicity of truncated PrPs.Citation14 These indicate that the neuroprotective activity of PrPC, especially mediated via the OR, may not be required for the antagonistic function of PrPC against Dpl.
Some models postulate that PrPC interacts with an as yet unidentified transmembrane molecule that transmits a neuroprotective or cell survival signal.Citation14,Citation18 Dpl and the toxic truncated PrPs could bind to the molecule, but generate no signal due to lack of the N-terminal domain, resulting in neuronal cell death. According to these models, PrP molecules that fail to generate the signal should be toxic, like Dpl or the toxic truncated PrPs. However, inconsistent with this, we previously showed that Construct 4 lacking residues 23–88, failed to elicit the antagonistic signal against Dpl but was itself non-toxic to neurons.Citation17
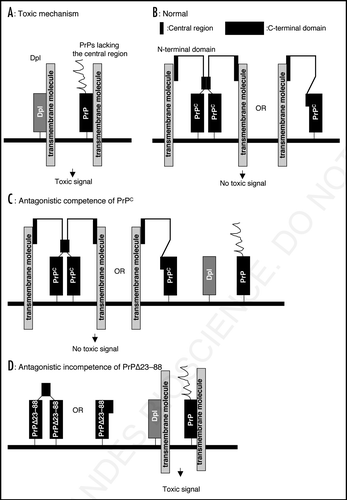
Wong et al. reported that Dpl-expressing Rcm0 Prnp0/0 mice produced oxidative stress of radical oxygen species or nitric oxide in their brains much more than non-expressing Npu Prnp0/0 mice.Citation28 This therefore suggested that Dpl may actively produce the neurotoxic signal, causing neuronal cell degeneration. Dpl is a GPI-anchored membrane glycoprotein, thus requiring interaction with a transmembrane molecule to transmit the signal (). The toxic PrP molecules may interact with the molecule via the Dpl-homologous C-terminal domain in the same way as Dpl, eliciting a neurotoxic signal (). However, the central region may interfere with the interaction, thereby preventing the neurotoxicity of the C-terminal of PrP in cis (). Rambold et al. reported that PrP with the domain spanning central residues 113–133 deleted, termed PrPΔHD, failed to form a homo-dimer, being toxic by inducing apoptosis in human neuroblastoma SH-SY5Y cells.Citation29 It is thus possible that the central residues may be involved in dimerization of PrP, thereby preventing the C-terminal domain of PrP from interaction with the transmembrane molecule (). Alternatively, the central residues may form intra-molecular interaction with the C-terminal region of PrP, thereby inhibiting the binding between it and the transmembrane molecule (). The N-terminal region which mediates the trans-protective activity, may also bind to the transmembrane molecule only when the cis-element is intact, but may produce no toxic signal (). The N-terminal region, together with the central region, may compete with Dpl or the truncated PrPs for the transmembrane molecule, resulting in reduction of the neurotoxicity of Dpl or the truncated PrPs in trans (). In contrast, PrP molecules which lack the N-terminal region, such as PrPΔ23–88, have no potential to protect against Dpl or the truncated PrPs in trans (). According to this model, the neurotoxicity of Dpl or the toxic PrPs is explained by interaction between Dpl or the Dpl-homologous C-terminal region of PrP and the putative transmembrane molecule (). The cis- and trans-neuroprotective activity may be mediated by disturbing the interaction in cis or trans via the central or N-terminal regions, respectively (). The neurotoxic PrP peptide, PrP106–126, and neurotoxic monoclonal anti-PrP antibodies, IgG D13 and P, which recognize an epitope (residues 95–105) very adjacent to the central region, may impair the cis-activity of the central region and promote the neurotoxic binding between the C-terminal region of PrPC and the transmembrane molecule, inducing neuronal cell death.Citation30,Citation31 However, this model can be verified only when the putative transmembrane molecule is identified.
Implication for Prion Diseases
Many lines of evidence indicate that conformational conversion of PrPC into the abnormally folded amyloidogenic isoform, PrPSc, plays a pivotal role in the pathogenesis of transmissible spongiform encephalopathies or prion diseases, including Creutzfeldt-Jakob disease in humans and bovine spongiform encephalopathy in cattle.Citation32 However, the molecular mechanism by which neurons undergo degenerative death remains unknown. PrPSc differs from PrPC in tertiary structure.Citation33 PrPC is rich in α-helix content while PrPSc have a markedly increased content of β-sheet.Citation33 Thus, due to the structural changes, the central region of PrPSc may lose its cis-activity and PrPSc therefore might interact with the putative transmembrane molecule, causing neuronal degeneration. Alternatively, association between PrPC and PrPSc during the structural conversion might impair the cis-activity of the associating PrPC, subsequently inducing neuronal cell death. N-terminally truncated forms of protease-resistant PrP have been reported to accumulate in the brains of patients affected with prion diseases and in persistently infected cultured cells.Citation34,Citation35 It may be also conceivable that these N-terminally truncated PrP fragments posses a neurotoxic potential equivalent to that of Dpl and Constructs 1 and 2 due to deletion of the cis-element. Thus, elucidation of a molecular mechanism of the antagonistic interaction between Dpl and PrPC could be useful for understanding of the molecular pathogenesis of prion diseases.
Figures and Tables
Figure 1 (A) Schemes of wild-type mouse PrP and Dpl. Mouse PrP is first translated as a precursor protein consisting of 254 amino acids. The N-terminal 22 and C-terminal 23 hydrophobic amino acids are removed as a signal peptide and a GPI-anchor signal sequence, respectively. The N-terminal half of PrPC is highly flexible and lacks identifiable secondary structure. The octapeptide repeat (OR) region, comprising five copies of a P(H/Q)GGG(G)WGQ octapeptide sequence, is located in the N-terminal domain. The OR region is thought to mediate anti-oxidative activity by binding to Cu2+ via histidine residues. However, the exact function of this region remains to be elucidated. The C-terminal half of PrPC forms a globular structure with three α-helices (α1–3) and two short anti-parallel β-strands (β1, β2). The second and third helices are linked by a disulfide bond (-S-S-). The precursor protein of Dpl consists of 179 amino acids. The N-terminal 25 and C-terminal 22 hydrophobic residues may be removed as signal peptide and GPI-anchor signals, respectively. Dpl is a structural homologue of the C-terminal globular domain of PrPC, sharing ∼23% identical amino acids and is composed of three α-helices (α1–3) and two short anti-parallel β-strands (β1, β2). Two disulfide bonds (-S-S-) are formed. However, Dpl lacks the corresponding N-terminal part of PrPC. (B) Structural schemes of PrPs with deletion of various regions and PrPN-Dpl, the fusion protein composed of the N-terminal region of PrP with Dpl, with their cis- and trans-protective activity against Dpl or toxic PrPs are shown. a: Construct 7 is itself non-toxic. However, it has different affects on neurotoxic Constructs 2 and 6: It enhances the toxicity of Construct 6 but diminishes that of Construct 2. NA: data are not available.
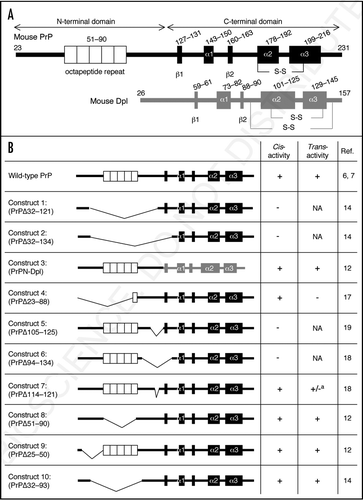
Figure 2 A possible mechanism for the antagonistic interaction of PrPC and Dpl or the toxic PrPs. (A) Dpl binds to a putative transmembrane molecule, producing a toxic signal. Toxic PrPs with deletion of the central region, such as Constructs 1, 2, 5 and 6, bind to the transmembrane molecule via the Dpl-homologous C-terminal area in the same way as Dpl, eliciting a similar toxic signal. (B) Under normal conditions, wild-type PrPC binds to the trans-membrane molecule via the N-terminal region but not its C-terminal region because it forms either a homo-dimer linked via the central region or a monomer with the central region interacting with part of the C-terminal domain. The N-terminal region acquires binding affinity to the molecule only when the central region is intact. However, this type of interaction produces no toxic signal. (C) PrPs with part of the N-terminal region and with the central region both intact, such as trans- protective PrPs, have a higher affinity for the transmembrane molecule than Dpl or the toxic PrPs, resulting in trans-protection against Dpl and the toxic PrPs. (D) Construct 4 (PrPΔ23–88) still has potential to form a homo-dimer due to the residual central region or a monomer with the residual central region masking part of the C-terminal region, similarly to wild-type PrPC. Therefore, Construct 4 cannot form a complex with the transmembrane molecule via the C-terminal region, generating no toxic signal. In addition, by lacking part of the N-terminal domain, Construct 4 has no affinity for the transmembrane molecule, losing trans-protective activity against Dpl.
Acknowledgements
This study is partly supported by a Research on Specific Diseases from the Ministry of Health, Labour and Welfare, Japan.
References
- Stahl N, Borchelt DR, Hsiao K, Prusiner SB. Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell 1987; 51:229 - 240
- Prusiner SB. Molecular biology of prion diseases. Science 1991; 252:1515 - 1522
- Oesch B, Westaway D, Walchli M, McKinley MP, Kent SB, Aebersold R, et al. A cellular gene encodes scrapie PrP 27–30 protein. Cell 1985; 40:735 - 746
- Moore RC, Lee IY, Silverman GL, Harrison PM, Strome R, Heinrich C, et al. Ataxia in prion protein (PrP)-deficient mice is associated with upregulation of the novel PrP-like protein doppel. J Mol Biol 1999; 292:797 - 817
- Li A, Sakaguchi S, Atarashi R, Roy BC, Nakaoke R, Arima K, et al. Identification of a novel gene encoding a PrP-like protein expressed as chimeric transcripts fused to PrP exon 1/2 in ataxic mouse line with a disrupted PrP gene. Cell Mol Neurobiol 2000; 20:553 - 567
- Moore RC, Mastrangelo P, Bouzamondo E, Heinrich C, Legname G, Prusiner SB, et al. Doppel-induced cerebellar degeneration in transgenic mice. Proc Natl Acad Sci USA 2001; 98:15288 - 15293
- Yamaguchi N, Sakaguchi S, Shigematsu K, Okimura N, Katamine S. Doppel-induced Purkinje cell death is stoichiometrically abrogated by prion protein. Biochem Biophys Res Commun 2004; 319:1247 - 1252
- Li A, Sakaguchi S, Shigematsu K, Atarashi R, Roy BC, Nakaoke R, et al. Physiological expression of the gene for PrP-like protein, PrPLP/Dpl, by brain endothelial cells and its ectopic expression in neurons of PrP-deficient mice ataxic due to Purkinje cell degeneration. Am J Pathol 2000; 157:1447 - 1452
- Rossi D, Cozzio A, Flechsig E, Klein MA, Rulicke T, Aguzzi A, et al. Onset of ataxia and Purkinje cell loss in PrP null mice inversely correlated with Dpl level in brain. EMBO J 2001; 20:694 - 702
- Sakaguchi S, Katamine S, Nishida N, Moriuchi R, Shigematsu K, Sugimoto T, et al. Loss of cerebellar Purkinje cells in aged mice homozygous for a disrupted PrP gene. Nature 1996; 380:528 - 531
- Yoshikawa D, Kopacek J, Yamaguchi N, Ishibashi D, Yamanaka H, Yamaguchi Y, et al. Newly established in vitro system with fluorescent proteins shows that abnormal expression of downstream prion protein-like protein in mice is probably due to functional disconnection between splicing and 3′ formation of prion protein pre-mRNA. Gene 2007; 386:139 - 146
- Yoshikawa D, Yamaguchi N, Ishibashi D, Yamanaka H, Okimura N, Yamaguchi Y, et al. Dominant-negative effects of the N-terminal half of prion protein on neurotoxicity of prion protein-like protein/doppel in mice. J Biol Chem 2008; 283:24202 - 24211
- Riek R, Hornemann S, Wider G, Glockshuber R, Wuthrich K. NMR characterization of the full-length recombinant murine prion protein, mPrP(23–231). FEBS Lett 1997; 413:282 - 288
- Shmerling D, Hegyi I, Fischer M, Blattler T, Brandner S, Gotz J, et al. Expression of amino-terminally truncated PrP in the mouse leading to ataxia and specific cerebellar lesions. Cell 1998; 93:203 - 214
- Anderson L, Rossi D, Linehan J, Brandner S, Weissmann C. Transgene-driven expression of the Doppel protein in Purkinje cells causes Purkinje cell degeneration and motor impairment. Proc Natl Acad Sci USA 2004; 101:3644 - 3649
- Mo H, Moore RC, Cohen FE, Westaway D, Prusiner SB, Wright PE, et al. Two different neurodegenerative diseases caused by proteins with similar structures. Proc Natl Acad Sci USA 2001; 98:2352 - 2357
- Atarashi R, Nishida N, Shigematsu K, Goto S, Kondo T, Sakaguchi S, et al. Deletion of N-terminal residues 23–88 from prion protein (PrP) abrogates the potential to rescue PrP-deficient mice from PrP-like protein/doppel-induced neurodegeneration. J Biol Chem 2003; 278:28944 - 28949
- Baumann F, Tolnay M, Brabeck C, Pahnke J, Kloz U, Niemann HH, et al. Lethal recessive myelin toxicity of prion protein lacking its central domain. EMBO J 2007; 26:538 - 547
- Li A, Christensen HM, Stewart LR, Roth KA, Chiesa R, Harris DA. Neonatal lethality in transgenic mice expressing prion protein with a deletion of residues 105–125. EMBO J 2007; 26:548 - 558
- McLennan NF, Brennan PM, McNeill A, Davies I, Fotheringham A, Rennison KA, et al. Prion protein accumulation and neuroprotection in hypoxic brain damage. Am J Pathol 2004; 165:227 - 235
- Sakurai-Yamashita Y, Sakaguchi S, Yoshikawa D, Okimura N, Masuda Y, Katamine S, et al. Female-specific neuroprotection against transient brain ischemia observed in mice devoid of prion protein is abolished by ectopic expression of prion protein-like protein. Neuroscience 2005; 136:281 - 287
- Weise J, Crome O, Sandau R, Schulz-Schaeffer W, Bahr M, Zerr I. Upregulation of cellular prion protein (PrPc) after focal cerebral ischemia and influence of lesion severity. Neurosci Lett 2004; 372:146 - 150
- Hoshino S, Inoue K, Yokoyama T, Kobayashi S, Asakura T, Teramoto A, et al. Prions prevent brain damage after experimental brain injury: a preliminary report. Acta Neurochir Suppl 2003; 86:297 - 299
- Kuwahara C, Takeuchi AM, Nishimura T, Haraguchi K, Kubosaki A, Matsumoto Y, et al. Prions prevent neuronal cell-line death. Nature 1999; 400:225 - 226
- Bounhar Y, Zhang Y, Goodyer CG, LeBlanc A. Prion protein protects human neurons against Bax-mediated apoptosis. J Biol Chem 2001; 276:39145 - 39149
- Mitteregger G, Vosko M, Krebs B, Xiang W, Kohlmannsperger V, Nolting S, et al. The role of the octarepeat region in neuroprotective function of the cellular prion protein. Brain Pathol 2007; 17:174 - 183
- Sakudo A, Lee DC, Nishimura T, Li S, Tsuji S, Nakamura T, et al. Octapeptide repeat region and N-terminal half of hydrophobic region of prion protein (PrP) mediate PrP-dependent activation of superoxide dismutase. Biochem Biophys Res Commun 2005; 326:600 - 606
- Wong BS, Liu T, Paisley D, Li R, Pan T, Chen SG, et al. Induction of HO-1 and NOS in doppel-expressing mice devoid of PrP: implications for doppel function. Mol Cell Neurosci 2001; 17:768 - 775
- Rambold AS, Muller V, Ron U, Ben-Tal N, Winklhofer KF, Tatzelt J. Stress-protective signalling of prion protein is corrupted by scrapie prions. Embo J 2008; 27:1974 - 1984
- Forloni G, Angeretti N, Chiesa R, Monzani E, Salmona M, Bugiani O, et al. Neurotoxicity of a prion protein fragment. Nature 1993; 362:543 - 546
- Solforosi L, Criado JR, McGavern DB, Wirz S, Sanchez-Alavez M, Sugama S, et al. Cross-linking cellular prion protein triggers neuronal apoptosis in vivo. Science 2004; 303:1514 - 1516
- Prusiner SB. Prions. Proc Natl Acad Sci USA 1998; 95:13363 - 13383
- Pan KM, Baldwin M, Nguyen J, Gasset M, Serban A, Groth D, et al. Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins. Proc Natl Acad Sci USA 1993; 90:10962 - 10966
- Chen SG, Teplow DB, Parchi P, Teller JK, Gambetti P, Autilio-Gambetti L. Truncated forms of the human prion protein in normal brain and in prion diseases. J Biol Chem 1995; 270:19173 - 19180
- Arima K, Nishida N, Sakaguchi S, Shigematsu K, Atarashi R, Yamaguchi N, et al. Biological and biochemical characteristics of prion strains conserved in persistently infected cell cultures. J Virol 2005; 79:7104 - 7112