Abstract
Classical bovine spongiform encephalopathy is a transmissible prion disease that is fatal to cattle and is a human health risk due to its association with a strain of Creutzfeldt-Jakob disease (vCJD). Mutations to the coding region of the prion gene (PRNP) have been associated with susceptibility to transmissible spongiform encephalopathies in mammals including bovines and humans. Additional loci such as the retinoic acid receptor beta (RARB) and stathmin like 2 (STMN2) have also been associated with disease risk. The objective of this study was to refine previously identified regions associated with BSE susceptibility and to identify positional candidate genes and genetic variation that may be involved with the progression of classical BSE. The samples included 739 samples of either BSE infected animals (522 animals) or non-infected controls (207 animals). These were tested using a custom SNP array designed to narrow previously identified regions of importance in bovine genome. Thirty one single nucleotide polymorphisms were identified at p < 0.05 and a minor allele frequency greater than 5%. The chromosomal regions identified and the positional and functional candidate genes and regulatory elements identified within these regions warrant further research.
Keywords: :
Introduction
Classical Bovine Spongiform Encephalopathy (BSE) is the variant of transmissible spongiform encephalopathies (TSEs) found in Bos taurus or bovines. The TSEs are progressive degenerative neurological disorders caused by the conversion of endogenous prion protein (PRPc) to an abnormally folded form (PrPRes)Citation1,Citation2 and are always fatal.Citation3 TSEs are unique in that they may be genetic, sporadic or transmitted but, in all cases the disease state is associated with accumulation of the abnormal PrPRes in the brain and central nervous system (CNS) causing degeneration.Citation4 Classical BSE is a form of TSE acquired by the consumption of meat and/or bone meal contaminated with the (PrPRes) or infectious prion agentCitation5 and was first observed in the United Kingdom (UK) in 1986.Citation6 A large number of infected animals entered the food chain in the preclinical state and were consumed by the human population in the UK and elsewhere.Citation7 This consumption has been linked to a variant form of Creutzfeldt-Jakob disease (vCJD)Citation8,Citation9 with approximately 200 reported cases in Europe , Asia, and North America (data from National Creutzfeldt-Jakob Disease Surveillance Unit (NCJDSU), Western General Hospital Edinburgh, Scotland: Worldwide vCJD statistics 2010).
Prion disease pathogenesis occurs when endogenous PrPRes auto catalyzes the conversion of PrPc into the PrPRes form. Exposure to the PrPRes promotes or facilitates this conversion through a mechanism that is not yet understood.Citation10 PrPRes is highly resistant to breakdown by the proteases and other proteolytic machinery of the cell.Citation11,Citation12 This resistance results in the accumulation of the non-degraded form of the protein which results in the neural degeneration and spongiform appearance of infected CNS tissues.Citation4 In classical BSE, the pathogenesis of the disease is the result of the transference of the infectious prion from the digestive tract into the peripheral nervous system and then to the brain.Citation5 Studies of the pathogenesis have concluded that the route of transmission is from the ileal Peyer’s patches and the tonsils through the parasympathetic and sympathetic nerve fibers of the autonomous nervous system.Citation5 It has been hypothesized that a number of cellular proteins and the genes may be involved in this progression.Citation13,Citation14 Structural variation in prion protein locus, the gene for the PrP protein, is strongly associated with the risk of all categories of prion disease in humans including vCJD.Citation15 In addition, specific PRNP alleles have been associated with TSE susceptibility in sheep.Citation16,Citation17 In classical BSE, variation in the promoter region of the PRNP locus has also been shown to be associated with risk of disease incidence.Citation18,Citation19 In addition to the PRNP locus, single nucleotide polymorphisms (SNPs) associated with other genes such as the retinoic acid receptor beta (RARB) and stathmin-like 2 proteins (STMN2) have been shown to be related to human susceptibility to prion disease.Citation15,Citation20 Genome wide association studies have also identified several other genomic regions associated with BSE incidenceCitation13,Citation14,Citation21,Citation22 but little is known about how these regions and the genes within them that may affect the pathogenesis of classical BSE. Additionally, genome wide association studies in humans have identified other loci of modest overall effect as research targets.Citation23 The objective of this study was to refine previously identified regions associated with BSE susceptibility and to identify positional candidate genes and genetic variation that may be involved with the progression of classical BSE.
Results and Discussion
Candidate gene identification
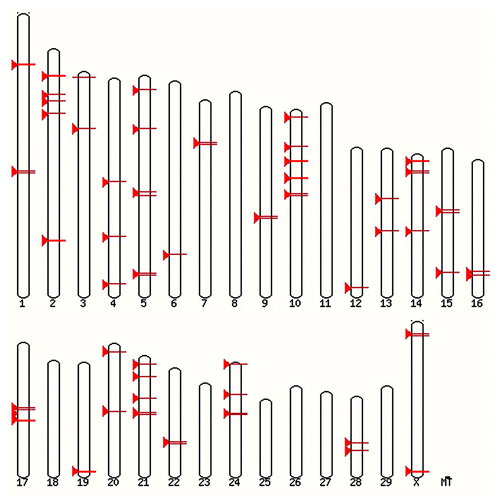
Fifty seven regions on BTAU 4.0Citation24 associated with BSE incidence and susceptibility were identified utilizing data generated previously within our lab and throughout published literature.Citation13,Citation14,Citation21,Citation22 These regions are shown in . A window of 1MB was created around each previously identified marker (500,000 bases to either side) in order to capture all of the genes closely linked to identified associations. Known genes within each 1MB pair window in BTAU 4.0 were then obtained using BIOMART.Citation25 All previously reported candidate genes for prion disease susceptibility,Citation21,Citation22 were included as well as newly identified candidate genes based on potential functional role in BSE. Candidate genes were identified using NCBI Genbank,Citation26 Gene OntologyCitation27 and KEGG PathwayCitation28 databases and were included if the gene was known to be expressed in nervous tissue, was involved in proteolysis, protein folding or unfolding or protein transport.
Figure 1. Regions of BTAU 4.0 previously identified as associated to incidence of classical BSE in cattle.
Marker identification for candidate genes and genome regions
Transcript sequences from liver, hypothalamus, muscle, adipose, duodenum, kidney, lung blood, cortex and Peyer’s patch were evaluated to identify SNPs in each of the 89 candidate genes mapped to the 57 regions of the bovine genome thought to be related to BSE susceptibility. Whole-genome sequences from a Holstein bull and a Black Angus bullCitation29were also used to identify structural variation within candidate genes. Non-synonymous mutations were selected when available as were SNPs predicted to introduce a stop codon or splice variation.
Overall structural variation was identified in 87 of 89 candidate genes. Additional SNPs were added to increase the resolution within the 57 one MB pair windows of BTAU 4.0.
The origin of all SNPs in the constructed panel is shown in Additional File 1. Whenever possible, markers were selected that were identified in more than one dataset.
Genotyping
A custom panel of 384 SNP markers was designed to evaluate 87 candidate genes identified in underlying QTLs for BSE incidence. The total number of SNP markers included in the analysis after quality control measures was 240. This panel was used to genotype 739 samples of either BSE infected animals (522 animals) or non-infected controls (207 animals). The animal samples came from a population of half-sibs and a set of unrelated case and control samples. The samples were combined for this study in a modified sib-TDT test using the DFAM procedure in the PLINK softwareCitation30 that allows for addition of unrelated individuals. The combined population was examined for stratification and none was observed.
A total of 31 markers were associated with incidence of BSE in this population at a significance level of p > 0.05 and a minor allele frequency greater than 5% (). Some of these SNPs are located close to one another and are potentially within LD in the population.
Table 1. Significance of associated marker loci using the DFAM Procedure in PLINK for a sib-TDT with unrelated individuals
Classical BSE as a phenotype
Previous studiesCitation21,Citation22 have suggested that it is difficult to test for the genetic association for the clinical presentation of BSE. The explanation provided is that this could be attributed to the fact that while animals that developed BSE are clearly susceptible to disease and the control samples may represent animals that were either not exposed to enough infectious material to develop disease or are in fact resistant to infection. This phenotypic variation may reduce the power to detect disease association. Also as noted,Citation21,Citation22 the use of stringent multiple testing corrections such as Bonferroni to this type of a phenotype may be overly prone to type II errors and thus discard real, though subtle associations in favor of reducing or eliminating type I or false positive results. In contrast, this study was designed to refine the previously identified resultsCitation13,Citation14,Citation21,Citation22by evaluating a reduced number of markers in a larger pooled population of animals. However, with the total of 240 markers included in the analysis and the criteria of p < 0.05 means that approximately 12 of the identified 31 markers may be significant by chance.
Regions previously identified for BSE susceptibility
This work utilized genomic regions previously identified or suggested to be important to classical BSE susceptibility. However, this work refined previous associations and identified novel candidate genes (). As shown in , the position of the associated markers in this study are close but not identical to those shown previously. The resolution of this work was also higher as the aim was to have markers every 100,000 base pairs in the target areas. The increased resolution allowed further refinement of candidate genes and the identification of regulatory regions that may impact disease susceptibility.
Table 2. Previously identified and currently refined genomic regions related to BSE susceptibility
New candidate genes
Several pathogenesis studies have concluded that the route of transmission of classical BSE is through consumption of the misfolded PRNP which is then transported into ileal Peyer’s patches and the tonsils and then through the parasympathetic and sympathetic nerve fibers of the autonomous nervous system.Citation5 The misfolded PRNP then leads to the misfolding of native PRNP, which resists degradation and accumulates in the CNS resulting in neurodegeneration.Citation5 It has been hypothesized that a number of cellular proteins and genes play an important role in the progression of the disease.Citation13,Citation14 Several proteins () are known to participate in transport of the prion protein across the gut epithelium, transport to dendritic cells, accumulation in lymph tissues and progression from lymph tissues to CNS tissues and specifically to the brain.
Table 3. Positional and functional candidate genes for BSE susceptibility
An example of a functional candidate gene that may be linked to the pathogenesis of classical BSE is NEDD8 (Neural precursor cell expressed, developmentally down-regulated 8), which is located at approximately 21.2CM on BTAU 10. It is close to a marker (10: 21273459) that was significant at p <0.005 and would likely be linked to that marker. NEDD8 encodes an 81 amino acid polypeptide which is 60% identical and 80% homologous to ubiquitin.Citation31 Amyloid precursor protein binding protein-1 (APP-BP1) binds to the carboxyl terminus of the amyloid precursor protein (APP) and serves as the activation enzyme for the ubiquitin-like protein, NEDD8.Citation32 NEDD8 conjugation pathway has shown to be essential for proteolytic targeting by ubiquitination.Citation33 Impairment of this process may contribute to the amyloid plaques evident in classical BSE. NEDD8 has been shown to accumulate in both glial and neuronal inclusions in several neurodegenerative conditions such as Parkinson disease, Alcoholic liver disease and Astrocytoma.Citation34 Co-location of APP-BP1 and NEDD8 is shown in lipid rafts in the hippocampus of Alzheimer’s patients but not in other less affected regions of the brain.Citation35 Further investigation is needed to validate a relationship between NEDD8 and to determine the nature of the potential relationship with classical BSE susceptibility.
Another important positional and functional candidate gene identified in this study is GDNF (glial cell derived neurotrophic factor). GDNF has been shown to mitigate neuronal degeneration in a range of neurodegenerative conditions and was one of the first investigated targets for gene therapy in Parkinson disease.Citation36,Citation37 GDNF is thought to promote survival and differentiation of developing neurons and to protect mature neurons.Citation36 Mutations in or impairments of GDNF action have been shown contribute to the symptoms of Down syndrome and Schizophrenia.Citation38 The action of GDNF could play a role in innate resistance to neurodegeneration in classical BSE. As with NEDD8, future research is needed to establish a relationship and to determine the potential role of GDNF as a target for BSE resistance.
Candidate markers near regulatory regions or CpG Islands
Several markers that were significantly associated with classical BSE susceptibility are positioned close to annotated CpG islands in the bovine genome. Vertebrate CpG islands are short DNA sequences that are significantly richer in GC bases as compared to the rest of the genome and are predominantly non-methylated.Citation39 Most, if not all, CpG islands are sites of transcription initiation though many are not currently annotated promotersCitation39. Shared DNA sequence features adapt CpG islands for promoter function by destabilizing nucleosomes and attracting proteins that create a transcriptionally preferable chromatin state.Citation39 The significantly associated SNP markers in close proximity to CpG islands identified in this study are shown in .
Table 4. Associated loci in a regulatory region and nearby candidate genes
One example of a candidate gene that could be relevant to classical BSE pathogenesis and is in close proximity to both a significant associated marker and CpG island is exostosin 1 (EXT1). As noted previously,Citation22 EXT1 is an endoplasmic reticulum transmembrane glycoprotein responsible for the synthesis and display of cell surface heparin sulphate glycosaminoglycans (GAGs).Citation40 The N terminus of PRNP contains a GAG-binding motif and that may play a functional role in the uptake and transport of aberrant PRNP in gastrointestinal and neural cells in classical BSE transmission and infection.Citation21,Citation41 This relationship needs to be evaluated in the bovine and the link to classical BSE disease needs to be shown in further research.
In conclusion, this research has identified the most exhaustive list of candidate genes for classical BSE susceptibility to date and has supported an association of 31 markers shown to be related to classical BSE susceptibility in a large dataset of 729 case and control animals. It has improved the resolution and power of previous analyses and provided strong functional and positional candidate genes which may lead to better understanding of this disease in its natural host and provide candidate gene and regulatory targets for further research.
Materials and Methods
Animal information
This study used two sets of animals including both case (affected) and control (unaffected) samples from two populations with different population structures. The first data set is family based and includes 225 BSE affected and 193 BSE unaffected half-sibs Holsteins from 30 sire families. These were paternal half-sibs with different dams from the UK which were collected during the BSE outbreak in the mid-1990s. The second population included 297 BSE affected and 14 BSE unaffected animals. The control animals are contemporaries of the cases and were collected from the same farms. In these two populations, BSE status was determined both by examination by qualified veterinarians and then post-mortem by histology (Veterinary Laboratories Agency, New Haw, Surrey, UK). The control animals are assumed to have been exposed to the same environment, did not exhibit any signs of disease and are assumed to have been disease free.
Genomic DNA was isolated from the family based population using a phenol chloroform extraction as described by Hernandez-Sanchez et al.Citation13 Genomic DNA from the case-control population was isolated using a high salt phenol/chloroform extraction as described by Sherman et al.Citation42
mRNA-seq library construction and sequencing using Illumina platform
RNA was prepared for liver (7 animals), hypothalamus (11 animals), muscle and duodenum (12 animals), kidney and lung (14 animals) and adipose (10 animals) tissues. Total RNA was extracted using TRIzol reagent (Invitrogen) from the frozen tissues. The quality and quantity of RNA was determined using a Nanodrop (Nanodrop technologies) and Agilent Bioanalyzer (Agilent).
The RNAs from each tissue were pooled and cDNA libraries were constructed from each tissue pool according to the protocol (Illumina). The first step in the process of Illumina library construction involved purifying the poly-A containing mRNA molecules from total RNA using poly-T oligo-attached magnetic beads. Poly (A) RNA was fragmented followed by random hexamer reverse transcription and second strand synthesis. These cDNA fragments were end repaired prior to ligation of the Illumina GEX PE adapters according to the standard library generation protocol (Illumina). A ~85 bp size range was excised from the library on 6% agarose gel and the resultant library was subjected to 15 cycles of PCR followed by quantification using Nanodrop (Nanodrop Technologies).
Sequencing was performed on Genome Analyzer II (Illumina) which uses a massively parallel sequencing-by-synthesis four-dye approach, to generate billions of bases of high-quality DNA sequence per run. Cluster generation and sequencing was performed on a PE 75nt X 75nt flow cell generating ~1.7-6.0 GB of sequence.
Maq (version 0.7.1) was used to map reads and to generate SNP lists.Citation43 First, the reads were converted from the Illumina export format to the Sanger FASTQ format using the “fq_all2std.pl export2std” command. The “match” command was used to align the paired-end reads to bovine transcripts from Ensembl (release 57).Citation44 The resulting alignments were then merged using the “mapmerge” command and consensus sequences were generated using the “assemble” command. The “cns2snp” command was used to build a list of potential SNP sites from the consensus sequences. A second shorter list of SNPs was also generated using the “maq.pl SNPfilter” command. Default settings were used in all cases, except that the -N option was used with the “assemble” command to specify the number of haplotypes present in each sample. The shorter, filtered SNP list was annotated using NGS-SNPCitation45 and release 57 of Ensembl,Citation44 which includes SNPs from dbSNP build 130.Citation46
Total RNA-Seq- library construction and sequencing using SOLiD platform
For each Cortex (8), Blood (10), Peyer’s Patch (6) libraries samples were pooled into 10ul before depleting the ribosomal RNA using the RiboMinus Eukaryote kit (Invitrogen) and concentrated with a RiboMinus Concentration Module. Five hundred nanograms (as determined by Agilent RNA Nano Chip on the Agilent Bioanalyzer) were carried through library prep using the whole transcriptome RNA-Seq Kit (Applied Biosystems). Libraries were quantified by QRTPCR using the Taqman kit (Applied Biosystems). Fragment sizes were checked using the DNA 1000 chip (Agilent) with the Agilent Bioanalyzer. Beads for Solid 4 were prepared using an EZBead system (Applied Biosystems). Sequencing was conducted on the SOLiD 4 system (ABI). Reads (50bp) were aligned to bovine transcripts from Ensembl (release 57)Citation44 and SNPs were identified by GEOSPIZA.
Whole genome sequencing of Angus and Holstein bull
Genomic DNA from a Black Angus bull and a Holstein bull was sequenced using the Applied Biosystems SOLiD 3 sequencer (Life Technologies Corporation), using a combination of fragment and mate-paired libraries. The libraries were prepared using the reagents and protocols provided by Applied Biosystems, and as reported in previous studies.Citation29
Sequence reads were mapped to the Btau4.0 bovine genome assembly using the Bioscope 1.0 software suite (Life Technologies Corporation). A list of putative SNPs was generated for each animal from the mapped reads, using the diBayes SNP Detection module (with the “med-coverage” stringency setting) included with Bioscope. The lists were subjected to additional filtering, to remove SNPs with particularly high read depth (higher than 95% of the other SNPs from the same animal), and to remove SNPs that could not be unambiguously placed on bovine genome assembly UMD3.1Citation47 using the megablast algorithmCitation48 in BLAST+,Citation49 100 bp of flanking sequence, and an e-value threshold of 1e-35. SNPs were annotated using NGS-SNPCitation45and release 57 of Ensembl,Citation44 which includes SNPs from dbSNP build 130.Citation46
Genotyping and data quality control
The total of 729 samples were genotyped with a custom Illumina Golden Gate Vera Code Assay containing 384 SNPs following the manufacturers recommended protocol (Illumina Inc.). Genotypes were called using the BeadStudio software (Illumina Inc.) and processed through the automated genotype calling. Genotypes were then subjected to data quality control parameters. Seventeen SNPs were removed for being monomorphic. Overall, a genotyping success rate of 96% was achieved. Forty seven SNPs from the original set of 384 SNPs were excluded on the basis of assay failure or poor genotype clustering. The remaining 320 SNPs were analyzed through the summary statistics of the PLINK program.Citation30 Additionally, 80 SNPs were removed due to a minor allele frequency (MAF) less than 0.01. In total 240 SNPs were included in the analysis.
Statistical analysis
PLINK software v1.07 was used to perform the statistical analysis.Citation30 The genotypic data from both populations was combined and was analyzed using the DFAM procedure in PLINK. The DFAM procedure in PLINK implements the sib-TDT and also allows for unrelated individuals to be included (via a clustered-analysis using the Cochran-Mantel-Haesnzel). This data was combined instead of being analyzed separately in the two populations due to the unbalanced nature of the second case-control population. This was the most extensive candidate gene list tested for BSE including more number of animals than previous work.
| Abbreviations: | ||
| BSE | = | bovine spongiform encephalopathy |
| PrP | = | prion protein |
| TSE | = | transmissible spongiform encephalopathy |
| vCJD | = | variant Creutzfeldt-Jakob disease |
| SNP | = | single nucleotide polymorphism |
| CNS | = | central nervous system |
Additional material
Download Zip (78.2 KB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgements
Jennifer Thomson constructed the custom marker panel, performed analysis, and wrote the manuscript; Victoria Bowles and Yan Meng participated in sequencing library construction; Urmila Basu contributed to the library construction, sequencing and manuscript writing; Victoria Bowles, Jung-Woo Choi contributed to SNP identification and marker panel construction, Paul Stothard and Stephen Moore were involved in experimental design, and manuscript writing.
Supplemental Material
Supplemental materials may be found here:
http://www.landesbioscience.com/journals/prion/article/21866/
References
- Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science 1982; 216:136 - 44; http://dx.doi.org/10.1126/science.6801762; PMID: 6801762
- Legname G, Baskakov IV, Nguyen HO, Riesner D, Cohen FE, DeArmond SJ, et al. Synthetic mammalian prions. Science 2004; 305:673 - 6; http://dx.doi.org/10.1126/science.1100195; PMID: 15286374
- Prusiner SB. Prions. Proc Natl Acad Sci U S A 1998; 95:13363 - 83; http://dx.doi.org/10.1073/pnas.95.23.13363; PMID: 9811807
- Novakofski J, Brewer MS, Mateus-Pinilla N, Killefer J, McCusker RH. Prion biology relevant to bovine spongiform encephalopathy. J Anim Sci 2005; 83:1455 - 76; PMID: 15890824
- Balkema-Buschmann A, Fast C, Kaatz M, Eiden M, Ziegler U, McIntyre L, et al. Pathogenesis of classical and atypical BSE in cattle. Prev Vet Med 2011; 102:112 - 7; http://dx.doi.org/10.1016/j.prevetmed.2011.04.006; PMID: 21592603
- Wells GA, Scott AC, Johnson CT, Gunning RF, Hancock RD, Jeffrey M, et al. A novel progressive spongiform encephalopathy in cattle. Vet Rec 1987; 121:419 - 20; http://dx.doi.org/10.1136/vr.121.18.419; PMID: 3424605
- Donnelly CA, Ferguson NM, Ghani AC, Anderson RM. Implications of BSE infection screening data for the scale of the British BSE epidemic and current European infection levels. Proc Biol Sci 2002; 269:2179 - 90; http://dx.doi.org/10.1098/rspb.2002.2156; PMID: 12427310
- Bruce ME, Will RG, Ironside JW, McConnell I, Drummond D, Suttie A, et al. Transmissions to mice indicate that ‘new variant’ CJD is caused by the BSE agent. Nature 1997; 389:498 - 501; http://dx.doi.org/10.1038/39057; PMID: 9333239
- Hill AF, Desbruslais M, Joiner S, Sidle KC, Gowland I, Collinge J, et al. The same prion strain causes vCJD and BSE. Nature 1997; 389:448 - 50, 526; http://dx.doi.org/10.1038/38925; PMID: 9333232
- Atarashi R, Moore RA, Sim VL, Hughson AG, Dorward DW, Onwubiko HA, et al. Ultrasensitive detection of scrapie prion protein using seeded conversion of recombinant prion protein. Nat Methods 2007; 4:645 - 50; http://dx.doi.org/10.1038/nmeth1066; PMID: 17643109
- Telling GC, Parchi P, DeArmond SJ, Cortelli P, Montagna P, Gabizon R, et al. Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Science 1996; 274:2079 - 82; http://dx.doi.org/10.1126/science.274.5295.2079; PMID: 8953038
- Riesner D. Biochemistry and structure of PrP(C) and PrP(Sc). Br Med Bull 2003; 66:21 - 33; http://dx.doi.org/10.1093/bmb/66.1.21; PMID: 14522846
- Hernández-Sánchez J, Waddington D, Wiener P, Haley CS, Williams JL. Genome-wide search for markers associated with bovine spongiform encephalopathy. Mamm Genome 2002; 13:164 - 8; http://dx.doi.org/10.1007/BF02684022; PMID: 11919688
- Zhang C, De Koning DJ, Hernández-Sánchez J, Haley CS, Williams JL, Wiener P. Mapping of multiple quantitative trait loci affecting bovine spongiform encephalopathy. Genetics 2004; 167:1863 - 72; http://dx.doi.org/10.1534/genetics.104.026401; PMID: 15342524
- Mead S, Poulter M, Uphill J, Beck J, Whitfield J, Webb TE, et al. Genetic risk factors for variant Creutzfeldt-Jakob disease: a genome-wide association study. Lancet Neurol 2009; 8:57 - 66; http://dx.doi.org/10.1016/S1474-4422(08)70265-5; PMID: 19081515
- Laegreid WW, Clawson ML, Heaton MP, Green BT, O’Rourke KI, Knowles DP. Scrapie resistance in ARQ sheep. J Virol 2008; 82:10318 - 20; http://dx.doi.org/10.1128/JVI.00710-08; PMID: 18632863
- Alvarez L, Arranz JJ, San Primitivo F. Identification of a new leucine haplotype (ALQ) at codon 154 in the ovine prion protein gene in Spanish sheep. J Anim Sci 2006; 84:259 - 65; PMID: 16424251
- Murdoch BM, Clawson ML, Yue S, Basu U, McKay S, Settles M, et al. PRNP haplotype associated with classical BSE incidence in European Holstein cattle. PLoS One 2010; 5:10.1371.; http://dx.doi.org/10.1371/journal.pone.0012786; PMID: 20862290
- Sander P, Hamann H, Drögemüller C, Kashkevich K, Schiebel K, Leeb T. Bovine prion protein gene (PRNP) promoter polymorphisms modulate PRNP expression and may be responsible for differences in bovine spongiform encephalopathy susceptibility. J Biol Chem 2005; 280:37408 - 14; http://dx.doi.org/10.1074/jbc.M506361200; PMID: 16141216
- Lloyd S, Mead S, Collinge J. Genetics of prion disease. Top Curr Chem 2011; 305:1 - 22; http://dx.doi.org/10.1007/128_2011_157; PMID: 21528440
- Murdoch BM, Clawson ML, Laegreid WW, Stothard P, Settles M, McKay S, et al. A 2cM genome-wide scan of European Holstein cattle affected by classical BSE. BMC Genet 2010; 11:20; http://dx.doi.org/10.1186/1471-2156-11-20; PMID: 20350325
- Murdoch BM, Murdoch GK, Settles M, McKay S, Williams JL, Moore SS. Genome-wide scan identifies loci associated with classical BSE occurrence. PLoS One 2011; 6:e26819; http://dx.doi.org/10.1371/journal.pone.0026819; PMID: 22073200
- Mead S, Uphill J, Beck J, Poulter M, Campbell T, Lowe J, et al. Genome-wide association study in multiple human prion diseases suggests genetic risk factors additional to PRNP. Hum Mol Genet 2012; 21:1897 - 906; http://dx.doi.org/10.1093/hmg/ddr607; PMID: 22210626
- Liu Y, Qin X, Song XZ, Jiang H, Shen Y, Durbin KJ, et al.. Bos taurus genome assembly. BMC Genomics 2009; 10:180.1471-2164-10-180
- Guberman JM, Ai J, Arnaiz O, Baran J, Blake A, Baldock R, et al. BioMart Central Portal: an open database network for the biological community. Database (Oxford) 2011
- Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Sayers EW. GenBank. Nucleic Acids Res 2011; 39:Database issue D32 - 7; http://dx.doi.org/10.1093/nar/gkq1079; PMID: 21071399
- Barrell D, Dimmer E, Huntley RP, Binns D, O’Donovan C, Apweiler R. The GOA database in 2009--an integrated Gene Ontology Annotation resource. Nucleic Acids Res 2009; 37:Database issue D396 - 403; http://dx.doi.org/10.1093/nar/gkn803; PMID: 18957448
- Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, Kanehisa M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res 1999; 27:29 - 34
- Stothard P, Choi JW, Basu U, Sumner-Thomson JM, Meng Y, Liao X, et al. Whole genome resequencing of black Angus and Holstein cattle for SNP and CNV discovery. BMC Genomics 2011; 12:559; http://dx.doi.org/10.1186/1471-2164-12-559; PMID: 22085807
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007; 81:559 - 75; http://dx.doi.org/10.1086/519795; PMID: 17701901
- Kim W, Bennett EJ, Huttlin EL, Guo A, Li J, Possemato A, et al. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol Cell 2011; 44:325 - 40; http://dx.doi.org/10.1016/j.molcel.2011.08.025; PMID: 21906983
- Kamitani T, Kito K, Nguyen HP, Yeh ET. Characterization of NEDD8, a developmentally down-regulated ubiquitin-like protein. J Biol Chem 1997; 272:28557 - 62; http://dx.doi.org/10.1074/jbc.272.45.28557; PMID: 9353319
- Podust VN, Brownell JE, Gladysheva TB, Luo RS, Wang C, Coggins MB, et al. A Nedd8 conjugation pathway is essential for proteolytic targeting of p27Kip1 by ubiquitination. Proc Natl Acad Sci U S A 2000; 97:4579 - 84; http://dx.doi.org/10.1073/pnas.090465597; PMID: 10781063
- Mori F, Nishie M, Piao YS, Kito K, Kamitani T, Takahashi H, et al. Accumulation of NEDD8 in neuronal and glial inclusions of neurodegenerative disorders. Neuropathol Appl Neurobiol 2005; 31:53 - 61; http://dx.doi.org/10.1111/j.1365-2990.2004.00603.x; PMID: 15634231
- Chen Y, Liu W, McPhie DL, Hassinger L, Neve RL. APP-BP1 mediates APP-induced apoptosis and DNA synthesis and is increased in Alzheimer’s disease brain. J Cell Biol 2003; 163:27 - 33; http://dx.doi.org/10.1083/jcb.200304003; PMID: 14557245
- Connor B, Dragunow M. The role of neuronal growth factors in neurodegenerative disorders of the human brain. Brain Res Brain Res Rev 1998; 27:1 - 39; http://dx.doi.org/10.1016/S0165-0173(98)00004-6; PMID: 9639663
- Grondin R, Gash DM. Glial cell line-derived neurotrophic factor (GDNF): a drug candidate for the treatment of Parkinson’s disease. J Neurol 1998; 245:Suppl 3 35 - 42; http://dx.doi.org/10.1007/PL00007744; PMID: 9808338
- Carter CJ. eIF2B and oligodendrocyte survival: where nature and nurture meet in bipolar disorder and schizophrenia?. Schizophr Bull 2007; 33:1343 - 53; http://dx.doi.org/10.1093/schbul/sbm007; PMID: 17329232
- Deaton AM, Bird A. CpG islands and the regulation of transcription. Genes Dev 2011; 25:1010 - 22; http://dx.doi.org/10.1101/gad.2037511; PMID: 21576262
- McCormick C, Leduc Y, Martindale D, Mattison K, Esford LE, Dyer AP, et al. The putative tumour suppressor EXT1 alters the expression of cell-surface heparan sulfate. Nat Genet 1998; 19:158 - 61; http://dx.doi.org/10.1038/514; PMID: 9620772
- Yin S, Pham N, Yu S, Li C, Wong P, Chang B, et al. Human prion proteins with pathogenic mutations share common conformational changes resulting in enhanced binding to glycosaminoglycans. Proc Natl Acad Sci U S A 2007; 104:7546 - 51; http://dx.doi.org/10.1073/pnas.0610827104; PMID: 17456603
- Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 1987; 162:156 - 9; http://dx.doi.org/10.1016/0003-2697(87)90021-2; PMID: 2440339
- Li H, Ruan J, Durbin R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res 2008; 18:1851 - 8; http://dx.doi.org/10.1101/gr.078212.108; PMID: 18714091
- Hubbard TJ, Aken BL, Ayling S, Ballester B, Beal K, Bragin E, et al. Ensembl 2009. Nucleic Acids Res 2009; 37:Database issue D690 - 7; http://dx.doi.org/10.1093/nar/gkn828; PMID: 19033362
- Grant JR, Arantes AS, Liao X, Stothard P. In-depth annotation of SNPs arising from resequencing projects using NGS-SNP. Bioinformatics 2011; 27:2300 - 1; http://dx.doi.org/10.1093/bioinformatics/btr372; PMID: 21697123
- Sherry ST, Ward M, Sirotkin K. dbSNP-database for single nucleotide polymorphisms and other classes of minor genetic variation. Genome Res 1999; 9:677 - 9; PMID: 10447503
- Zimin AV, Delcher AL, Florea L, Kelley DR, Schatz MC, Puiu D, et al. A whole-genome assembly of the domestic cow, Bos taurus. Genome Biol 2009; 10:R42; http://dx.doi.org/10.1186/gb-2009-10-4-r42; PMID: 19393038
- Zhang Z, Schwartz S, Wagner L, Miller W. A greedy algorithm for aligning DNA sequences. J Comput Biol 2000; 7:203 - 14; http://dx.doi.org/10.1089/10665270050081478; PMID: 10890397
- Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, et al. BLAST+: architecture and applications. BMC Bioinformatics 2009; 10:421; http://dx.doi.org/10.1186/1471-2105-10-421; PMID: 20003500
- Shieh JT, Swidler P, Martignetti JA, Ramirez MC, Balboni I, Kaplan J, et al. Systemic hyalinosis: a distinctive early childhood-onset disorder characterized by mutations in the anthrax toxin receptor 2 gene (ANTRX2). Pediatrics 2006; 118:e1485 - 92; http://dx.doi.org/10.1542/peds.2006-0824; PMID: 17043134
- Potkin SG, Turner JA, Guffanti G, Lakatos A, Fallon JH, Nguyen DD, et al, FBIRN. A genome-wide association study of schizophrenia using brain activation as a quantitative phenotype. Schizophr Bull 2009; 35:96 - 108; http://dx.doi.org/10.1093/schbul/sbn155; PMID: 19023125
- Joo Y, Ha S, Hong BH, Kim J, Chang KA, Liew H, et al. Amyloid precursor protein binding protein-1 modulates cell cycle progression in fetal neural stem cells. PLoS One 2010; 5:e14203; http://dx.doi.org/10.1371/journal.pone.0014203; PMID: 21151996
- Ravikumar B, Rubinsztein DC. Role of autophagy in the clearance of mutant huntingtin: a step towards therapy?. Mol Aspects Med 2006; 27:520 - 7; http://dx.doi.org/10.1016/j.mam.2006.08.008; PMID: 16973207
- Rubinsztein DC. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 2006; 443:780 - 6; http://dx.doi.org/10.1038/nature05291; PMID: 17051204
- Lee JA, Liu L, Javier R, Kreitzer AC, Delaloy C, Gao FB. ESCRT-III subunits Snf7-1 and Snf7-2 differentially regulate transmembrane cargos in hESC-derived human neurons. Mol Brain 2011; 4:37; http://dx.doi.org/10.1186/1756-6606-4-37; PMID: 21975012
- Zenker M, Mayerle J, Lerch MM, Tagariello A, Zerres K, Durie PR, et al. Deficiency of UBR1, a ubiquitin ligase of the N-end rule pathway, causes pancreatic dysfunction, malformations and mental retardation (Johanson-Blizzard syndrome). Nat Genet 2005; 37:1345 - 50; http://dx.doi.org/10.1038/ng1681; PMID: 16311597
- Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 2007; 447:661 - 78; http://dx.doi.org/10.1038/nature05911; PMID: 17554300
- Airavaara M, Pletnikova O, Doyle ME, Zhang YE, Troncoso JC, Liu QR. Identification of novel GDNF isoforms and cis-antisense GDNFOS gene and their regulation in human middle temporal gyrus of Alzheimer disease. J Biol Chem 2011; 286:45093 - 102; http://dx.doi.org/10.1074/jbc.M111.310250; PMID: 22081608
- Larance M, Kirkwood KJ, Xirodimas DP, Lundberg E, Uhlen M, Lamond AI. Characterization of MRFAP1 turnover and interactions downstream of the NEDD8 pathway. Mol Cell Proteomics 2012; 11:M111 - , 014407; http://dx.doi.org/10.1074/mcp.M111.014407; PMID: 22038470
- Li H, Wetten S, Li L, St Jean PL, Upmanyu R, Surh L, et al. Candidate single-nucleotide polymorphisms from a genomewide association study of Alzheimer disease. Arch Neurol 2008; 65:45 - 53; http://dx.doi.org/10.1001/archneurol.2007.3; PMID: 17998437
- Hafler DA, Compston A, Sawcer S, Lander ES, Daly MJ, De Jager PL, et al, International Multiple Sclerosis Genetics Consortium. Risk alleles for multiple sclerosis identified by a genomewide study. N Engl J Med 2007; 357:851 - 62; http://dx.doi.org/10.1056/NEJMoa073493; PMID: 17660530
- Fung HC, Scholz S, Matarin M, Simón-Sánchez J, Hernandez D, Britton A, et al. Genome-wide genotyping in Parkinson’s disease and neurologically normal controls: first stage analysis and public release of data. Lancet Neurol 2006; 5:911 - 6; http://dx.doi.org/10.1016/S1474-4422(06)70578-6; PMID: 17052657
- Grupe A, Li Y, Rowland C, Nowotny P, Hinrichs AL, Smemo S, et al. A scan of chromosome 10 identifies a novel locus showing strong association with late-onset Alzheimer disease. Am J Hum Genet 2006; 78:78 - 88; http://dx.doi.org/10.1086/498851; PMID: 16385451
- Wang Q, Sharma D, Ren Y, Fondell JD. A coregulatory role for the TRAP-mediator complex in androgen receptor-mediated gene expression. J Biol Chem 2002; 277:42852 - 8; http://dx.doi.org/10.1074/jbc.M206061200; PMID: 12218053
- Rienzo M, Nagel J, Casamassimi A, Giovane A, Dietzel S, Napoli C. Mediator subunits: gene expression pattern, a novel transcript identification and nuclear localization in human endothelial progenitor cells. Biochim Biophys Acta 2010; 1799:487 - 95; http://dx.doi.org/10.1016/j.bbagrm.2010.05.001; PMID: 20493979
- Kim BT, Kitagawa H, Tamura J, Saito T, Kusche-Gullberg M, Lindahl U, et al. Human tumor suppressor EXT gene family members EXTL1 and EXTL3 encode alpha 1,4- N-acetylglucosaminyltransferases that likely are involved in heparan sulfate/ heparin biosynthesis. Proc Natl Acad Sci U S A 2001; 98:7176 - 81; http://dx.doi.org/10.1073/pnas.131188498; PMID: 11390981
- Nadanaka S, Kitagawa H. Heparan sulphate biosynthesis and disease. J Biochem 2008; 144:7 - 14; http://dx.doi.org/10.1093/jb/mvn040; PMID: 18367479
- Warner RG, Hundt C, Weiss S, Turnbull JE. Identification of the heparan sulfate binding sites in the cellular prion protein. J Biol Chem 2002; 277:18421 - 30; http://dx.doi.org/10.1074/jbc.M110406200; PMID: 11882649