Abstract
Upon prion infection, abnormal prion protein (PrPSc) self-perpetuate by conformational conversion of α-helix-rich PrPC into β sheet enriched form, leading to formation and deposition of PrPSc aggregates in affected brains. However the process remains poorly understood at the molecular level and the regions of PrP critical for conversion are still debated. Minimal amino acid substitutions can impair prion replication at many places in PrP. Conversely, we recently showed that bona fide prions could be generated after introduction of eight and up to 16 additional amino acids in the H2-H3 inter-helix loop of PrP. Prion replication also accommodated the insertions of an octapeptide at different places in the last turns of H2. This reverse genetic approach reveals an unexpected tolerance of prions to substantial sequence changes in the protease-resistant part which is associated with infectivity. It also demonstrates that conversion does not require the presence of a specific sequence in the middle of the H2-H3 area. We discuss the implications of our findings according to different structural models proposed for PrPSc and questioned the postulated existence of an N- or C-terminal prion domain in the protease-resistant region.
Self-Propagation of a Pathogenic Protein: Conversion of PrP into Different Prion Structures or Assemblies
Prions are the etiologic agents of transmissible fatal neurodegenerative diseases affecting both men and animals.Citation1 They are mainly if not solely composed of assemblies of PrPSc, a conformationally altered isoform of the host-encoded cellular prion protein PrPC. This PrPC is a glycoprotein tethered at the cell surface by a GPI-anchor. The N-terminal region of the protein is unstructured while the C-terminal moiety is a globular domain containing three α helices and two small anti-parallel β strands.Citation2-Citation4 In contrast, PrPSc is enriched in β structure, insoluble and tends to aggregate.Citation5-Citation7 Upon infection, exogenous PrPSc seeds are thought to self-template host PrPC, leading to further aggregation and deposition mainly in the nervous tissue. PrPC expression is essential for prion replication as shown by the resistance of PrP knockout mice to prion infection and the restoration of their susceptibility after introduction of a PrP transgene.Citation8,Citation9 Within the same host species, different prion strains can be propagated; those can be differentiated on the basis of the incubation time to disease, the pathology and PrPSc biochemical signature. Strain properties are assumed to be enciphered within differences in PrPSc conformation, at the level of the tertiary and/or quaternary structure.Citation10,Citation11
PrPSc was initially differentiated from PrPC by its insolubility and resistance to proteases such as proteinase K (PK).Citation1 While a strain-dependent, variable proportion of PrPSc is now recognized as PK-sensitive,Citation12-Citation15 its relative degree of infectivity is still debated. For most prion strains, the upstream N-terminal part of PrPSc is truncated following proteinase K (PK) treatment while the segment encompassing the 140 to 150 C-terminal amino acids shows high PK resistance. Similarly, in vivo, part of PrPSc is internalized and subjected to an N-terminal truncation mediated by endo-lysosomal enzymes.Citation16,Citation17
Natural Variation and Experimental Modification in ihe Sequence of Prions
The susceptibility of many mammalian species to experimental or ‘natural’ prion infection indicates that distinct, yet conserved PrP sequences are competent for prion conversion. Aligning the amino acid sequence of PrP PK-resistant core from mouse, hamster, human and ovine species () shows that differences can occur all along this segment. While the last 4 or 5 C-terminal amino acids preceding the GPI anchor attachment site are less conserved among species, they do not appear to be essential for prion replication.Citation18,Citation19 Apart from this C-terminus, the majority of the differences shown in are considered as equivalent replacement by comparison softwares such as BLAST. However, their combination is sufficient to potentially establish a species barrier to foreign prions.Citation20,Citation21 Oppositely, prions can cross the species barrier when adequate mutations are introduced in the PrP sequence of a given host-species. For example, substitutions in the N-terminal moiety of mouse PK-resistant segment, by residues found at the same place in hamster, human or sheep PrP render the transgenic mice expressing these chimeric proteins permissive to hamster, human and sheep prions respectively.Citation22-Citation24 These findings, along the pivotal role of the Met/Val polymorphism at PrP codon 129 in the human susceptibility to prions,Citation25 suggested that the region upstream from the last two helices is a major domain involved in the conformational change. The successful, cell-free amplification of a PK-resistant entity from the short PrP segment corresponding to the disease-associated human mutation Y145Stop recently further highlighted the potential role of this region.Citation26 However many familial forms of prion diseases are associated with mutations downstream position 145 of human PrP sequence. The C-terminal part of the protein may thus also play an important role in the conformational change.
Figure 1. Sequence alignment of the human, mouse, hamster and sheep PK-resistant PrPSc peptide. Numbering was according to the human sequence. Letters in blue correspond to amino acids present in the main PK fragment of 21 kDa strains (or Type 1) but not in 19 kDa (or Type 2) ones. Letters highlighted in red indicate difference with the human sequence and those highlighted in yellow indicate amino acids considered as similar (or positive) using the blast comparison software.

While PrP with deletions within the protease resistant domain may induce non-transmissible neuropathology in transgenic mice,Citation27-Citation29 those are not permissive to prions. The only exception was observed with the so called miniprion PrP106, i.e., PrP with two deletions (23–88 and 141–176).Citation30 The 141–176 region was nevertheless suggested to participate in the structural changes leading to PrPSc.Citation31,Citation32 Recently PrPs with myc or tetracysteine tags inserted at the end of the PK-resistant domain were shown to be convertible.Citation19,Citation33,Citation34 To further gain insight into the critical region(s) of the PrP sequence involved in the conformational change leading to PrPSc, we generated a panel of PrP mutants with an insertion inside the PK-resistant core and tested their ability of conversion into prions in cell culture.
Prions with Insertions in the Middle of PK-Resistant Core
We have recently shown that bona fide prions could be generated following insertion of ectopic peptides in H2 end or in the H2-H3 inter-helix loop of ovine PrPC.Citation35 These mutant proteins were stably expressed in RK13 cells, a rabbit kidney cell line permissive to 127S ovine prions, upon expression of ovine PrP (V136R154Q171 haplotype).Citation36,Citation37 The mutant PrP were converted by 127S prions and self-propagated PrPSc through multiple cell passages. The mutant cells with the original insert (, first lane) were as infectious as their wild-type counterparts, for recipient cells expressing either the homologous insertion mutant or the wild-type PrP and for transgenic mice expressing ovine PrPVRQ. Other inserts were introduced at the same place. They were eight to 16 amino acids long and some contained a polyglycine stretch, a His-tag, or a FLAG-tag ( and ).Citation35 All accommodated 127S prion replication, indicating that prion conversion did not require a specified size, a specific sequence, or a defined amino acid composition in the H2-H3 loop. While this tolerance to sequence change was exceptional, it has limits as introduction of a tetracysteine-tag renders PrP resistant to conversion (). This finding might be connected with a previous study, where mouse PrP with a tetracysteine tag introduced into the same loop did not convert into fibrils in vitro.Citation38 F and T residues conservation in front of H3 might be necessary to efficient conversion as these amino acids were kept in our initial study, so as to minimize the eventual impacts on the formation of the last helix. Their removal from an otherwise well tolerated insert indeed impaired prion replication (, compare lanes 1 and 8 or lanes 5 and 9). Accordingly, substitution in wild-type PrP of the F or the T residues of the NFT glycosylation site was not always compatible with prion conversionCitation36 (see also ). Another salient finding was that prion conversion also accommodated insertion of as much as 8 additional amino acids in the C-terminal region of H2 (). The octapeptide was introduced upstream of the third residue from the C-terminus of H2 and further upstream with gradual amino acid increment. Four consecutive mutants PrP were converted into PrPSc upon 127S infection. Nonetheless insertion before the sixth residue from H2 end dramatically reduced conversion efficiency and insertions further upstream were not converted anymore ().
Table 1. Changes introduced in ovine PrPC sequence and their compatibility with prion conversion
Figure 2. Insertion site of peptides compatible with PrPSc generation. The 3D structure of sheep PrP is shown with the sequence of peptides inserted in the inter-helix loop, i.e., in between the NFT glycosylation site and the beginning of helix α 3. Dark letters correspond to amino acid of the original insert isolated, red ones to engineered modifications and blue ones to FLAG-tag sequence introduced either inside the original peptide as published,Citation35 or directly inserted at position 203 (lane below, not previously published).
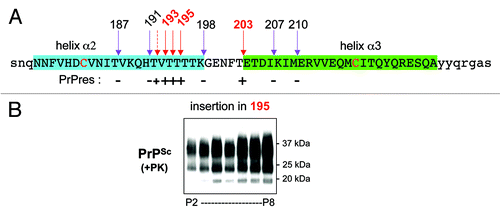
Figure 3. Displacement of the octapeptide insert (AQQGGFT) in H2-H3. (A) Segments corresponding to H2 and H3 are in blue and in green, respectively. Cysteines of the disulfide bridge are identified in red. Arrows show the position of the insertions, red arrows indicate compatibility with prion replication and purple ones incompatibility, as indicated by positive or negative production of self-propagating PK-resistant PrPSc (PrPres) as shown on the line below the sequence. (B) western blot analysis of PK-digested samples of PrP-ins195 from cell culture passages two to eight following 127S strain infection.
To determine whether such insertions modified PrPC structure, the same changes were introduced in PrP expressed in E. coli, assuming that the 3D structures of these recombinant proteins would be equivalent to those of the cell-membrane anchored glycoproteins and would provide relevant information.Citation39 Circular dichroism analysis did not reveal significant modification of the secondary structure of PrP with an octapeptide inserted either at position 193 (convertible, see ) or two amino acids upstream (unconvertible). Because a slight decrease in the proportion of alpha-helical content may be difficult to detect with such technique, more detailed structural analysis have been initiated by NMR. HSQC recordings indicate that the insertions did not alter PrP global arrangement. It might suggest that some of the amino acids inserted have replaced those of the helix.
Together our findings reveal the high tolerance of prions to major sequence modifications in the C-terminal part of H2 and adjacent loop of PrP. This conclusion was further sustained by the convertibility of a double mutant PrP with two octapeptide inserts, one at the end of H2 and the other in the inter helix loop, right in front of H3.Citation35
Insertions in the H2-H3 Domain and Structural Models of Prpsc
While high-resolution structure of PrPSc is lacking,Citation40 several models have been proposed in which the participation of H2-H3 domain is debated.Citation32,Citation41-Citation44 The β helixCitation31,Citation32 and the in-register modelsCitation44 have in common to propose the existence of a “prion domain” (as in yeast prions)Citation45 shorter than the whole PK-resistant segment, but they conflictingly locate it in the N or C-terminal part, respectively. The tolerance of prions to sequence change in the middle of H2-H3 region, either in the inter-helix segment or in the C-terminal part of H2, tends to exclude the middle part of H2-H3 as the main prion conversion site. Our results also strongly suggest that the inter-helix segment remains essentially unstructured in PrPSc because insertion of peptide not prone to form secondary structures, such as the polyglycine stretch can be tolerated at that place. However they do not exclude that the conversion process transform H3 and most of H2 into β strands. Indeed insertions in H3, or upstream to the last turns of H2 were not compatible with prion conversion, while these mutant PrP were normally expressed at the cell surface.
The existence of a small prion domain might itself be questioned. How the conformationally unchanged part outside such domain would resist protease digestion? It was long believed that there were 15% to 30% alpha-helical structures in PrPSc, suggesting that the C-terminal part of PrP remained essentially unmodified in the PK-resistant core while the N-terminal half underwent a conformational change. Recent FT-IR spectroscopic data and re-interpretation of earlier works might challenge this view, as they lead to the conclusion that there is no more α-helix in PrPSc.Citation5,Citation46 On the other hand, a putative C-terminal prion domain emerged from structural analysis of recombinant PrP polymers.Citation42,Citation44 To accommodate these data, it might worth considering that the prion domain is congruent with the whole protease-resistant domain. In this respect some theoretical models such as formation of both an N and a C-terminal left hand β helix or domain swapping of the molecules, were also proposed.Citation41,Citation43 Establishment of the 3D structure of PrPSc arrangement as well as definition of the landscape of sequence elements critical for conversion and those susceptible to be modified remains a major challenge to fully understand the conversion process and identify drug targets.
| Abbreviations: | ||
| PrP | = | prion protein |
| PrPC | = | normal cellular PrP |
| PrPSc | = | scrapie associated PrP |
| PK | = | proteinase K |
| PrPres | = | PK-resistant PrPSc |
| GPI | = | glycophosphatidylinositol |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Analisa Pastore (NIMR London, UK) and Stephanie Prigent (INRA Jouy-en-Josas, France) for their help with recombinant PrP structural analysis. We also thank Jerôme Chapuis for help with mutagenesis and Laetitia Hertzog and Emilie Jaumain for analysis of infected mice. This work was supported by INRA. M. K. F. Salamat was supported by the Higher Education Commission of Pakistan and by INRA Animal Health division.
Related Research Data
References
- Prusiner SB. Prions. Proc Natl Acad Sci U S A 1998; 95:13363 - 83; http://dx.doi.org/10.1073/pnas.95.23.13363; PMID: 9811807
- Eghiaian F, Grosclaude J, Lesceu S, Debey P, Doublet B, Tréguer E, et al. Insight into the PrPC-->PrPSc conversion from the structures of antibody-bound ovine prion scrapie-susceptibility variants. Proc Natl Acad Sci U S A 2004; 101:10254 - 9; http://dx.doi.org/10.1073/pnas.0400014101; PMID: 15240887
- James TL, Liu H, Ulyanov NB, Farr-Jones S, Zhang H, Donne DG, et al. Solution structure of a 142-residue recombinant prion protein corresponding to the infectious fragment of the scrapie isoform. Proc Natl Acad Sci U S A 1997; 94:10086 - 91; http://dx.doi.org/10.1073/pnas.94.19.10086; PMID: 9294167
- Riek R, Hornemann S, Wider G, Billeter M, Glockshuber R, Wüthrich K. NMR structure of the mouse prion protein domain PrP(121-231). Nature 1996; 382:180 - 2; http://dx.doi.org/10.1038/382180a0; PMID: 8700211
- Baron GS, Hughson AG, Raymond GJ, Offerdahl DK, Barton KA, Raymond LD, et al. Effect of glycans and the glycophosphatidylinositol anchor on strain dependent conformations of scrapie prion protein: improved purifications and infrared spectra. Biochemistry 2011; 50:4479 - 90; http://dx.doi.org/10.1021/bi2003907; PMID: 21539311
- Pan KM, Baldwin M, Nguyen J, Gasset M, Serban A, Groth D, et al. Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins. Proc Natl Acad Sci U S A 1993; 90:10962 - 6; http://dx.doi.org/10.1073/pnas.90.23.10962; PMID: 7902575
- Caughey BW, Dong A, Bhat KS, Ernst D, Hayes SF, Caughey WS. Secondary structure analysis of the scrapie-associated protein PrP 27-30 in water by infrared spectroscopy. Biochemistry 1991; 30:7672 - 80; http://dx.doi.org/10.1021/bi00245a003; PMID: 1678278
- Fischer M, Rülicke T, Raeber A, Sailer A, Moser M, Oesch B, et al. Prion protein (PrP) with amino-proximal deletions restoring susceptibility of PrP knockout mice to scrapie. EMBO J 1996; 15:1255 - 64; PMID: 8635458
- Büeler H, Aguzzi A, Sailer A, Greiner RA, Autenried P, Aguet M, et al. Mice devoid of PrP are resistant to scrapie. Cell 1993; 73:1339 - 47; http://dx.doi.org/10.1016/0092-8674(93)90360-3; PMID: 8100741
- Aguzzi A, Heikenwalder M, Polymenidou M. Insights into prion strains and neurotoxicity. Nat Rev Mol Cell Biol 2007; 8:552 - 61; http://dx.doi.org/10.1038/nrm2204; PMID: 17585315
- Peretz D, Williamson RA, Legname G, Matsunaga Y, Vergara J, Burton DR, et al. A change in the conformation of prions accompanies the emergence of a new prion strain. Neuron 2002; 34:921 - 32; http://dx.doi.org/10.1016/S0896-6273(02)00726-2; PMID: 12086640
- D’Castro L, Wenborn A, Gros N, Joiner S, Cronier S, Collinge J, et al. Isolation of proteinase K-sensitive prions using pronase E and phosphotungstic acid. PLoS One 2010; 5:e15679; http://dx.doi.org/10.1371/journal.pone.0015679; PMID: 21187933
- Colby DW, Wain R, Baskakov IV, Legname G, Palmer CG, Nguyen HO, et al. Protease-sensitive synthetic prions. PLoS Pathog 2010; 6:e1000736; http://dx.doi.org/10.1371/journal.ppat.1000736; PMID: 20107515
- Cronier S, Gros N, Tattum MH, Jackson GS, Clarke AR, Collinge J, et al. Detection and characterization of proteinase K-sensitive disease-related prion protein with thermolysin. Biochem J 2008; 416:297 - 305; http://dx.doi.org/10.1042/BJ20081235; PMID: 18684106
- Safar J, Wille H, Itri V, Groth D, Serban H, Torchia M, et al. Eight prion strains have PrP(Sc) molecules with different conformations. Nat Med 1998; 4:1157 - 65; http://dx.doi.org/10.1038/2654; PMID: 9771749
- Dron M, Moudjou M, Chapuis J, Salamat MK, Bernard J, Cronier S, et al. Endogenous proteolytic cleavage of disease-associated prion protein to produce C2 fragments is strongly cell- and tissue-dependent. J Biol Chem 2010; 285:10252 - 64; http://dx.doi.org/10.1074/jbc.M109.083857; PMID: 20154089
- Caughey B, Raymond GJ, Ernst D, Race RE. N-terminal truncation of the scrapie-associated form of PrP by lysosomal protease(s): implications regarding the site of conversion of PrP to the protease-resistant state. J Virol 1991; 65:6597 - 603; PMID: 1682507
- Scott MR, Safar J, Telling G, Nguyen O, Groth D, Torchia M, et al. Identification of a prion protein epitope modulating transmission of bovine spongiform encephalopathy prions to transgenic mice. Proc Natl Acad Sci U S A 1997; 94:14279 - 84; http://dx.doi.org/10.1073/pnas.94.26.14279; PMID: 9405603
- Taguchi Y, Shi ZD, Ruddy B, Dorward DW, Greene L, Baron GS. Specific biarsenical labeling of cell surface proteins allows fluorescent- and biotin-tagging of amyloid precursor protein and prion proteins. Mol Biol Cell 2009; 20:233 - 44; http://dx.doi.org/10.1091/mbc.E08-06-0635; PMID: 18987338
- Béringue V, Herzog L, Jaumain E, Reine F, Sibille P, Le Dur A, et al. Facilitated cross-species transmission of prions in extraneural tissue. Science 2012; 335:472 - 5; http://dx.doi.org/10.1126/science.1215659; PMID: 22282814
- Collinge J, Clarke AR. A general model of prion strains and their pathogenicity. Science 2007; 318:930 - 6; http://dx.doi.org/10.1126/science.1138718; PMID: 17991853
- Kupfer L, Eiden M, Buschmann A, Groschup MH. Amino acid sequence and prion strain specific effects on the in vitro and in vivo convertibility of ovine/murine and bovine/murine prion protein chimeras. Biochim Biophys Acta 2007; 1772:704 - 13; http://dx.doi.org/10.1016/j.bbadis.2006.10.009; PMID: 17145171
- Telling GC, Scott M, Hsiao KK, Foster D, Yang SL, Torchia M, et al. Transmission of Creutzfeldt-Jakob disease from humans to transgenic mice expressing chimeric human-mouse prion protein. Proc Natl Acad Sci U S A 1994; 91:9936 - 40; http://dx.doi.org/10.1073/pnas.91.21.9936; PMID: 7937921
- Scott M, Groth D, Foster D, Torchia M, Yang SL, DeArmond SJ, et al. Propagation of prions with artificial properties in transgenic mice expressing chimeric PrP genes. Cell 1993; 73:979 - 88; http://dx.doi.org/10.1016/0092-8674(93)90275-U; PMID: 8098995
- Collinge J, Palmer MS, Dryden AJ. Genetic predisposition to iatrogenic Creutzfeldt-Jakob disease. Lancet 1991; 337:1441 - 2; http://dx.doi.org/10.1016/0140-6736(91)93128-V; PMID: 1675319
- Abdallah A, Wang P, Richt JA, Sreevatsan S. Y145Stop is sufficient to induce de novo generation prions using protein misfolding cyclic amplification. Prion 2012; 6:81 - 8; http://dx.doi.org/10.4161/pri.6.1.18493; PMID: 22453182
- Baumann F, Tolnay M, Brabeck C, Pahnke J, Kloz U, Niemann HH, et al. Lethal recessive myelin toxicity of prion protein lacking its central domain. EMBO J 2007; 26:538 - 47; http://dx.doi.org/10.1038/sj.emboj.7601510; PMID: 17245436
- Li A, Christensen HM, Stewart LR, Roth KA, Chiesa R, Harris DA. Neonatal lethality in transgenic mice expressing prion protein with a deletion of residues 105-125. EMBO J 2007; 26:548 - 58; http://dx.doi.org/10.1038/sj.emboj.7601507; PMID: 17245437
- Shmerling D, Hegyi I, Fischer M, Blättler T, Brandner S, Götz J, et al. Expression of amino-terminally truncated PrP in the mouse leading to ataxia and specific cerebellar lesions. Cell 1998; 93:203 - 14; http://dx.doi.org/10.1016/S0092-8674(00)81572-X; PMID: 9568713
- Supattapone S, Bosque P, Muramoto T, Wille H, Aagaard C, Peretz D, et al. Prion protein of 106 residues creates an artifical transmission barrier for prion replication in transgenic mice. Cell 1999; 96:869 - 78; http://dx.doi.org/10.1016/S0092-8674(00)80596-6; PMID: 10102274
- Wille H, Bian W, McDonald M, Kendall A, Colby DW, Bloch L, et al. Natural and synthetic prion structure from X-ray fiber diffraction. Proc Natl Acad Sci U S A 2009; 106:16990 - 5; http://dx.doi.org/10.1073/pnas.0909006106; PMID: 19805070
- Govaerts C, Wille H, Prusiner SB, Cohen FE. Evidence for assembly of prions with left-handed beta-helices into trimers. Proc Natl Acad Sci U S A 2004; 101:8342 - 7; http://dx.doi.org/10.1073/pnas.0402254101; PMID: 15155909
- Goold R, Rabbanian S, Sutton L, Andre R, Arora P, Moonga J, et al. Rapid cell-surface prion protein conversion revealed using a novel cell system. Nat Commun 2011; 2:281; http://dx.doi.org/10.1038/ncomms1282; PMID: 21505437
- Rutishauser D, Mertz KD, Moos R, Brunner E, Rülicke T, Calella AM, et al. The comprehensive native interactome of a fully functional tagged prion protein. PLoS One 2009; 4:e4446; http://dx.doi.org/10.1371/journal.pone.0004446; PMID: 19209230
- Salamat K, Moudjou M, Chapuis J, Herzog L, Jaumain E, Béringue V, et al. Integrity of helix 2-helix 3 domain of the PrP protein is not mandatory for prion replication. J Biol Chem 2012; 287:18953 - 64; http://dx.doi.org/10.1074/jbc.M112.341677; PMID: 22511770
- Salamat MK, Dron M, Chapuis J, Langevin C, Laude H. Prion propagation in cells expressing PrP glycosylation mutants. J Virol 2011; 85:3077 - 85; http://dx.doi.org/10.1128/JVI.02257-10; PMID: 21248032
- Vilette D, Andreoletti O, Archer F, Madelaine MF, Vilotte JL, Lehmann S, et al. Ex vivo propagation of infectious sheep scrapie agent in heterologous epithelial cells expressing ovine prion protein. Proc Natl Acad Sci U S A 2001; 98:4055 - 9; http://dx.doi.org/10.1073/pnas.061337998; PMID: 11259656
- Gaspersic J, Hafner-Bratkovic I, Stephan M, Veranic P, Bencina M, Vorberg I, et al. Tetracysteine-tagged prion protein allows discrimination between the native and converted forms. FEBS J 2010; 277:2038 - 50; http://dx.doi.org/10.1111/j.1742-4658.2010.07619.x; PMID: 20345906
- Hornemann S, Schorn C, Wüthrich K. NMR structure of the bovine prion protein isolated from healthy calf brains. EMBO Rep 2004; 5:1159 - 64; http://dx.doi.org/10.1038/sj.embor.7400297; PMID: 15568016
- Requena JR. Structure of mammalian prions. Future Virology 2009; 4:295 - 307; http://dx.doi.org/10.2217/fvl.09.8
- Hafner-Bratkovič I, Jerala R. Disulfide mapping reveals the domain swapping as the crucial process of the structural conversion of prion protein. Prion 2011; 5:56 - 9; http://dx.doi.org/10.4161/pri.5.2.16232; PMID: 21555920
- Chakroun N, Prigent S, Dreiss CA, Noinville S, Chapuis C, Fraternali F, et al. The oligomerization properties of prion protein are restricted to the H2H3 domain. FASEB J 2010; 24:3222 - 31; http://dx.doi.org/10.1096/fj.09-153924; PMID: 20410442
- Kunes KC, Clark SC, Cox DL, Singh RR. Left handed beta helix models for mammalian prion fibrils. Prion 2008; 2:81 - 90; http://dx.doi.org/10.4161/pri.2.2.7059; PMID: 19098440
- Cobb NJ, Sönnichsen FD, McHaourab H, Surewicz WK. Molecular architecture of human prion protein amyloid: a parallel, in-register beta-structure. Proc Natl Acad Sci U S A 2007; 104:18946 - 51; http://dx.doi.org/10.1073/pnas.0706522104; PMID: 18025469
- Wickner RB. Discovering protein-based inheritance through yeast genetics. J Biol Chem 2012; 287:14432 - 42; http://dx.doi.org/10.1074/jbc.X112.355636; PMID: 22396539
- Smirnovas V, Baron GS, Offerdahl DK, Raymond GJ, Caughey B, Surewicz WK. Structural organization of brain-derived mammalian prions examined by hydrogen-deuterium exchange. Nat Struct Mol Biol 2011; 18:504 - 6; http://dx.doi.org/10.1038/nsmb.2035; PMID: 21441913