Abstract
Transmissible spongiform encephalopathies (TSEs) are fatal neurodegenerative diseases that are based on the misfolding of a cellular prion protein (PrPC) into an infectious, pathological conformation (PrPSc). There is proof-of-principle evidence that a prion vaccine is possible but this is tempered with concerns of the potential dangers associated with induction of immune responses to a widely-expressed self-protein. By targeting epitopes that are specifically exposed upon protein misfolding, our group developed a vaccine that induces PrPSc-specific antibody responses. Here we consider the ability of this polyclonal antibody (SN6b) to bind to a mutant of PrPC associated with spontaneous prion disease. Polyclonal antibodies were selected to mimic the vaccination outcome and also explore all possible protein conformations of the recombinant bovine prion protein with mutation T194A [bPrP(T194A)]. This mutant is a homolog of the human T183A mutation of PrPC that is associated with early onset of familial dementia. With nanopore analysis, under non-denaturing conditions, we observed binding of the SN6b antibody to bPrP(T194A). This interaction was confirmed through ELISAs as well as immunoprecipitation of the recombinant and cellularly expressed forms of bPrP(T194A). This interaction did not promote formation of a protease resistant conformation of PrP in vitro. Collectively, these findings support the disease-specific approach for immunotherapy of prion diseases but also suggest that the concept of conformation-specific immunotherapy may be complicated in individuals who are genetically predisposed to PrPC misfolding.
Introduction
Prion diseases of domestic animals include scrapie in sheep and goats, bovine spongiform encephalopathy (BSE) in cattle and chronic wasting disease (CWD) in deer and elk.Citation1 Human forms of prion disease include Creutzfeldt-Jakob disease (CJD), Gerstmann-Sträussler-Scheinker syndrome (GSS) and fatal familial insomnia (FFI).Citation2,Citation3 These diseases all share a progressive, fatal outcome as there are currently no effective treatment strategies. The “protein-only” hypothesis of prion diseases states that infectivity resides in the pathologically misfolded prion protein designated PrPSc. PrPSc is hypothesized to serve as a template to recruit the normal cellular protein (PrPC) to the infectious conformation through an autocatalytic process.Citation4 Interestingly, the concept of infectivity based on protein misfolding was once the exclusive domain and defining characteristic of the prion diseases, this model of self-propagation is now being considered for a number of other protein misfolding diseases including amyotrophic lateral sclerosis (ALS), Alzheimer disease and Parkinson disease.Citation5-Citation9
The majority of PrPSc transmission within animal populations is thought to occur through oral routes by environmental contamination or recycling of animal proteins in the food chain.Citation1 Efforts to eliminate prion transmission within cattle have focused on removal of animal protein from ruminant feeds.Citation10 The ongoing, although drastically reduced, incidence of BSE likely reflects a spontaneous form of the disease known as atypical BSE.Citation11 While the efforts to control BSE have been successful, management of prion diseases in wild populations of animals is far more complex. CWD has been detected in free ranging cervids in 17 US states and the Canadian provinces of Saskatchewan and Alberta. CWD is now considered as one of the most contagious TSEs that can reach a prevalence of 30% in wild populations and as high as 100% in captive cervids.Citation12,Citation13 The inability of currently available disease management tools to control the spread of CWD in wild populations of cervids has prompted efforts for the development of new tools for disease management, in particular a vaccine.
Considerable effort has gone into the development of a prion vaccine. A number of research groups have reported on vaccines that have utilized different epitopes, strategies of formulation and delivery, routes and timings of administration.Citation1,Citation14 While these efforts have failed to generate an effective vaccine, they have provided, based upon varying degrees of protection or delay of onset, proof-of-principle evidence that a vaccine may ultimately be an achievable goal for the control of prion diseases. The majority of these efforts have been vaccines that induce immune responses that fail to discriminate PrPC from PrPSc such that autoreactive antibodies to the normal, properly folded form of the prion protein are observed. There is concern that therapeutic targeting of PrPC—a virtually ubiquitous, cell surface protein may have adverse consequences.Citation14 Specifically, the systemic presence of autoreactive PrPC antibodies could compromise the natural function of PrPC, resulting in inappropriate cell signal activation, or stimulate suppressor T-cell lymphocytes.Citation15 It has been demonstrated that antibodies which crosslink PrPC in neural tissue induce apoptosisCitation16 and our group has demonstrated that antibody binding to PrPC alters patterns of intracellular signal transduction in human neurons.Citation17 In contrast, the induction of an immune response which is specific for the PrPSc conformation circumvents autoreactive PrPC antibodies and minimizes the potential adverse effects of prion immunoprophylaxis.
The most crucial step in the development of immunotherapies for prion diseases is to overcome self-tolerance to endogenous PrPC.Citation18 On the other hand PrPSc-specific immunotherapy is dependent on the identification and application of vaccine targets (epitopes) that are uniquely revealed upon protein misfolding. These disease-specific epitopes (DSEs) represent strong candidate vaccine targets for their potential to induce conformation-specific immune responses that spare the function of the properly folded protein. One such epitope, YYR, was identified for its unique exposure upon PrPC misfolding.Citation19 The lack of YYR-specific antibody reactivity with PrPC or other cell surface proteins, suggests that prion immunoprophylaxis may be possible with minimum risk of altering characteristics of PrPC. With the YYR epitope as a starting point, our group translated this weakly immunogenic epitope into a vaccine that induces robust and consistent PrPSc-specific-immune responses in a range of species. Specifically, through dual optimization of the epitope sequence and length, as well as strategies of formulation and delivery, we generated a vaccine that induces consistent and sustained serum PrPSc-specific IgG antibody responses with no consequences to the health and viability of the vaccinated animals.Citation20 This vaccine has considerable potential as a tool to manage animal prion diseases, in particular for chronic wasting disease where rates of exposure of elk and deer to infectious prions are at a crisis.
In contrast to the rapid progression of CWD through cervid populations the threat of prion diseases to human health is currently relatively low. Sporadic (sCJD), which lacks an obvious genetic component, is currently the most common form of human prion disease.Citation21 It is currently unknown whether sCJD results from endogenous or exogenous factors.Citation22 Familial prion diseases account for about 5‒15% of human TSE occurrences.Citation23,Citation24 This includes familial Creutzfeldt-Jakob disease (CJD), which occurs at a rate of 1 in a million people/year, GSS at a rate of less than 5 in 100 million people/year and fatal familial insomnia, which has only been characterized in 50 families.Citation2,Citation25,Citation26 There are a number of mutations of the prion protein gene (PRNP) that are associated with disease states.Citation27 For example, an inherited prion disease in humans is associated with the mutation T183A.Citation28,Citation29 This mutation has been suggested to disrupt glycosylation,Citation28 promote structural instability and misfoldingCitation30,Citation31 and accelerate conversion of the α-helical to β-sheet rich conformation.Citation32 Independent of the specific mechanism, this mutation correlates with early onset spontaneous prion disease.Citation28,Citation29 The low occurrence of prion diseases in the human population makes it highly unlikely that a prion vaccine would ever be administered to the general population. Instead, such a vaccine might be strategically administered to high risk individuals, such as those working with prions or be utilized as a therapeutic vaccine for persons genetically predisposed to prion diseases. The application of such a vaccine to genetically at-risk subjects requires further efforts to characterize the binding proclivities of the antibodies within the context of these specific PrPC sequence variations.
In the present work we investigate the ability of a PrPSc-specific antibody (SN6b) to interact with a mutant of PrPC, bPrP(T194A), associated with spontaneous prion disease. Nanopore analysis is used as one of the primary techniques to investigate the potential interaction. Through nanopore analysis, a change in current is observed when single molecules interact with an α-hemolysin pore embedded in a lipid membrane.Citation33,Citation34 Each molecule is interrogated individually, and thus, several conformations in the same sample can be analyzed simultaneously as long as each gives rise to distinct event parameters, namely the blockade current (I) and blockade time (T). Through nanopore analysis it was found that the SN6b antibody binds mutant bPrP(T194A), but not wild-type bPrP, under non-denaturing conditions. These findings were validated by ELISAs as well as immunoprecipitation of mutant protein both in the context of the bacterially expressed protein as well as that which is expressed within eukaryotic cells. In addition to the concerns that the antibody-PrP interaction may alter the function of the PrPC protein to the detriment of the host, there is the potential that the antibody, in combination with a misfolding prone PrPC, could function as a molecular chaperone to promote/stabilize the PrPSc conformation, thus inducing disease. The mutant recombinant protein alone or with the SN6b antibody, however, did not show formation of a protease resistant species. Overall, these findings support the safety of the PrPSc-specific approach to immunotherapy within high risk species, such as elk and deer, but suggest that the concept of PrPSc specificity may be compromised within individuals who are genetically predisposed to prion diseases. The significance of this interaction to the health and viability of the organism is not clear at this time. This does raise potentially interesting questions, however, to the concept of disease-specific immunotherapy for protein misfolding diseases. Specifically, individuals with genetic susceptibilities for these disorders may represent the highest priority, as well as the greatest challenge, for the application of these approaches.
Results
Nanopore analysis
Antibody prion interactions were analyzed using an α-hemolysin pore as described previously.Citation34,Citation35 In nanopore analysis, generally, type-I events have small blockade currents and short blockade times and correspond to individual molecules bumping into the pore before rapidly diffusing away.Citation36 Type-II events have larger blockade currents and longer blockade times and correspond to either translocation of a molecule through the pore after unfolding or transient insertion of a loop or strand of the protein into the pore before diffusing back to the cis side.Citation36 This latter type of event has been called intercalation and, for proteins it is often difficult to distinguish between translocation and intercalation.Citation36 Therefore, in this paper we will simply refer to events as either type-I (bumping) or type-II (translocation/intercalation). As shown previously, the events centered between these type-I and type-II may represent peptide/protein molecules that interact with the pore either by orientation through N- or C-terminal entry or by different conformations.Citation35 At present these possibilities cannot be distinguished. Previously, our group has used nanopore analysis to probe various aspects of PrPC behavior, including metal bindingCitation37 and the interaction with PrPC specific- antibodies.Citation35 As well, it was demonstrated that the event profiles after pre-incubation in 1.5 M guanidinium hydrochloride (Gdn-HCl) of wild-type bPrP and mutant bPrP(T194A) were different as were those of full length human PrP(23–231) and truncated PrP(90–231).Citation38 Thus the electronic signal at the instant that the protein interacts with the pore is very sensitive to the protein’s structure and conformation.Citation39-Citation41
A complete profile for wild-type bovine prion protein bPrP, could be accumulated in about 3 h (). The complete event parameters are listed (). There is a Gaussian distribution centered at -70 pA, which is due to type-II events, and a peak at -30 pA which is typical of bumping events (type-I). As shown previously, the proportion of Type-I events are about 60%.Citation37 Upon addition of antibody SN6b there was no significant change in the percentage of type-I events as would be expected if the antibody did not bind to the native protein (). For bPrP pre-treated with 1 M Gdn-HCl there was a significant increase in type-II events (84%) and a concomitant decrease in type-I events (16%) (). Upon the addition of SN6b, after treating with 1 M Gdn-HCl, there was a significant increase in the number of type-1 events to 38% (). Thus, the partial unfolding of PrP by Gdn-HCl, allowed the antibody access to its epitope.
Figure 1. Nanopore analysis demonstrates E.coli expressed bovine PrPC does not interact with antibody SN6b in the absence of Gdn-HCl. Current blockade histograms for bPrP and antibody SN6b. (A) bPrP, (B) bPrP with SN6b at a 1:1 ratio, (C) bPrP pre-treated in 1M Gdn-HCl, (D) bPrP pre-treated in 1M Gdn-HCl with SN6b at a 1:1 ratio. The values are listed in .
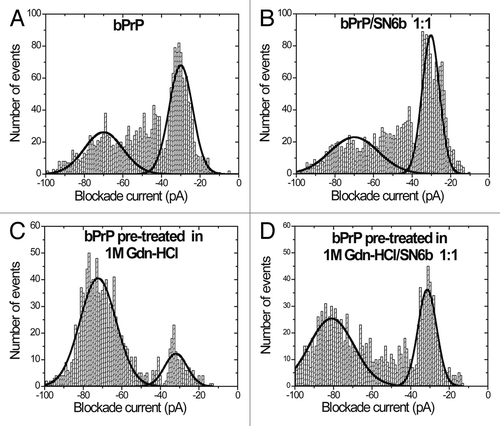
Table 1. Event parameters for bPrP pre-treated with 0M, 1M Gdn-HCl, bPrP(0M Gdn-HCl)/SN6b complex and bPrP(1M Gdn-HCl)/SN6b complex a
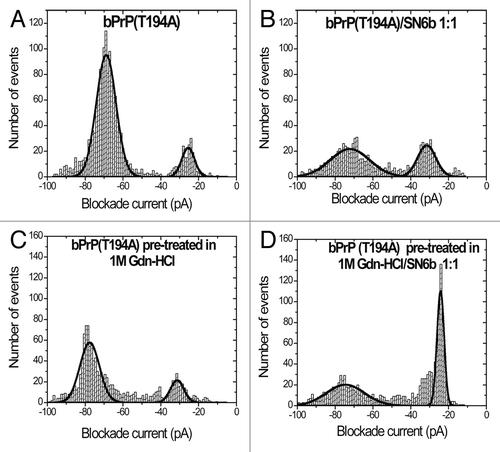
The event profiles for bPrP(T194A) are easily distinguishable from the wild-type (). Type-II events now predominate (87%) and the Gaussian distribution is much sharper (). Thus, as previously described for other proteins, such as the 86 amino acid HPr or the 140 amino acid α-synuclein, single amino acid substitutions cause subtle conformational changes which are readily detected by the pore.Citation40,Citation42 Upon addition of SN6b antibody, however, the proportion of type-II events decreases to 65% demonstrating that some of the native protein has a conformation that exposes the SN6b epitope (). When bPrP(T194A) is pre-treated with 1 M Gdn-HCl there is a small decrease in the number of type-II events in contrast to the effect of Gdn-HCl on the wild-type protein (). Upon addition of antibody SN6b the majority of events are type-I (51%) demonstrating binding to the partially unfolded protein ().
Table 2. Event parameters for bPrP(T194A) pre-treated with 0M, 1M, bPrP(T194A)(0M Gdn-HCl)/SN6b complex and bPrP(T194A)(1M Gdn-HCl)/SN6b complexa
Figure 2. Nanopore analysis demonstrates E.coli expressed bovine T194A mutant of PrPC interacts with antibody SN6b in the absence of Gdn-HCl. Current blockade histograms for bPrP(T194A) and antibody SN6b. (A) bPrP(T194A), (B) bPrP(T194A) with SN6b at a 1:1 ratio, (C) bPrP(T194A) pre-treated in 1M Gdn-HCl, (D) bPrP(T194A) pre-treated in 1M Gdn-HCl with SN6b at a 1:1 ratio. The values are listed in .
It was previously shown that the antibody M2188 binds wild-type bPrPCitation35 and confirmed that pre-treatment with 1M Gdn-HCl does not interfere with this interaction (, ). M2188 epitope exposure for wild-type bPrP appears to be independent of the conformational changes induced by Gdn-HCl. Similarly, at a protein-to-immunoglobulin G (IgG) ratio of 1:1, M2188 binds well to bPrP(T194A) after partial unfolding (1M Gdn-HCl) with almost 100% type-I events (, ). Surprisingly, the non-denatured protein binds M2188 less efficiently with only 58% type-I events (, ). Thus, there are subtle conformational differences between wild-type and mutant proteins both before and after treatment with Gdn-HCl. Additionally, nanopore analyses with antibody 6D11 in the absence of Gdn-HCl was conducted with the T194A mutant protein. On addition of 6D11 to bPrP(T194A) at a protein-to-immunoglobulin G (IgG) ratio of 4:1 the number of type-I events increased by 38% with a corresponding decrease of type-II events (, ). Finally, as a negative control, treatment of bPrP(T194A) with anti-FLAG antibody has no significant effect on the event profiles (compare and ).
Figure 3. Nanopore analysis demonstrates E.coli expressed bovine PrPC and T194A mutant of PrPC interacts with antibody M2188 in the presence of Gdn-HCl. Current blockade histograms for (A) bPrP pre-treated in 1M Gdn-HCl with antibody M2188 at a 1:1 ratio and (B) bPrP(T194A) pre-treated in 1M Gdn-HCl, with antibody M2188 at a 1:1 ratio. The values are listed in .
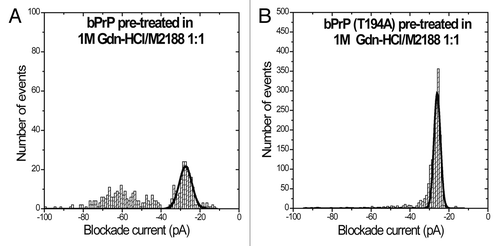
Table 3. Event parameters for bPrP(1M Gdn-HCl)/M2188 complex and bPrP(T194A)(1M Gdn-HCl)/M2188 complexa
Figure 4. Nanopore analysis in the absence of Gdn-HCl demonstrates, E.coli expressed T194A mutant of bovine PrPC interacts with antibodies M2188 and 6D11 that were used as positive controls, but not with anti-FLAG used as a negative control. Current blockade histograms for bPrP(T194) with antibodies M2188, 6D11 and anti-FLAG in the absence of Gdn-HCl. (A) bPrP(T194A) with antibody M2188 at a 1:1 ratio. (B) bPrP(T194A) with antibody 6D11 at a 4:1 ratio. (C) bPrP(T194A) with antibody anti-FLAG at a 2:1 ratio. The values are listed in .
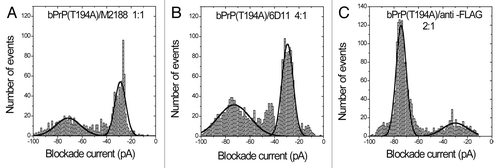
Table 4. Event parameters for bPrP(T194A)(0M Gdn-HCl)/M2188 complex, bPrP(T194A)(0M Gdn-HCl)/6D11 complex and bPrP(T194A)(0M Gdn-HCl)/anti-FLAG complexa
Immunoprecipitation of recombinant wild-type and T194A bovine PrPC
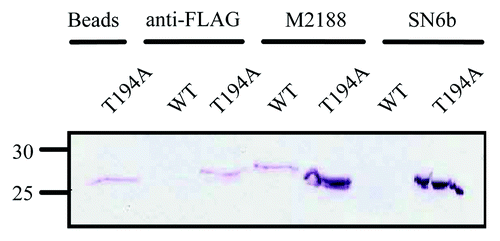
The potential interactions between the various antibodies with the wild-type bPrP and bPrP(T194A) proteins was considered in more conventional binding assays. Pull down assays with wild-type and T194A bovine proteins were conducted with SN6b, M2188 and anti-FLAG antibodies. Western blot analysis demonstrated that SN6b coupled beads immunoprecipitated bPrP(T194A) but not wild-type PrP. In contrast, the M2188 antibody immunoprecipitated both wild-type bPrP and bPrP(T194A) proteins (). Anti-FLAG coupled beads did not bind to wild-type bPrP but pulled down a small quantity of bPrP(T194A) which was interpreted to be background non-specific binding to the beads, as uncoupled beads were able to bind a similar amount (). Thus, while the T194A protein could bind non-specifically to the beads, both the SN6b and M2188 antibodies bound quantities of this protein well above background binding levels ().
Figure 5. Immunoprecipitation of E. coli expressed T194A bPrPC, but not wild-type bPrPC by SN6b coupled beads. The T194A mutant of bPrPC is immunoprecipitated with antibody uncoupled beads. Wild-type bovine PrPC is not pulled down by anti-FLAG antibody (negative control) as compared with bPrP(T194A) which is immunoprecipitated with anti-FLAG antibody with a similar band intensity as that obtained with the uncoupled beads. Bovine PrPC or T194A of bPrPC is immunoprecipitated with antibody M2188 (positive control), in which T194A shows a protein band that has intensity higher when compared with the band for PrPC. Bovine PrPC is not immunoprecipitated with antibody SN6b as compared with T194A mutant of bPrPC that is immunoprecipitated, depicting a band of similar intensity as that of M2188 with T194A. Molecular size markers in kDa are indicated on the left hand side of the panel.
ELISAs of recombinant wild-type and T194A bovine PrPC
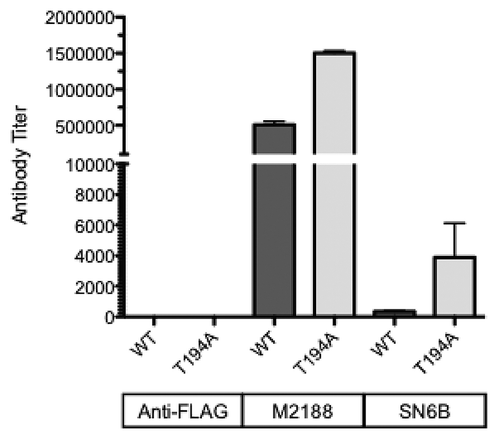
ELISAs confirmed the preferential binding of the SN6b antibody to bPrP(T194A), whereas titers observed for M2188 against wild-type and T194A proteins were similar (). No titer was observed for the negative control anti-FLAG antibody against either protein suggesting ELISA to be more specific than the immunoprecipitation assays for the E. coli. expressed recombinant prion proteins.
Figure 6. SN6b specificity in ELISAs. Affinity purified sheep SN6b was assessed for reactivity against both wild-type and T194A of bovine PrPC in ELISAs. The M2188 PrPC-specific monoclonal antibody and anti-FLAG antibody were included as positive and negative controls, respectively.
Immunoprecipitation of wild-type and T194A of PrPC expressed in HEK293 cells
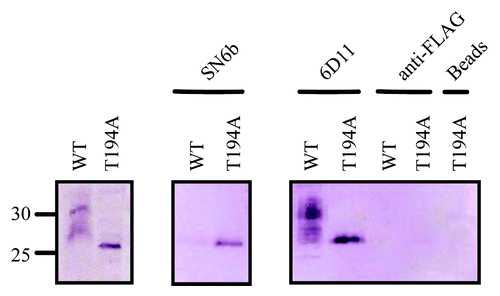
As PrPC is subject to in vivo post-translational modifications absent in prokaryotes, it was important to determine whether the observed differences in SN6b antibody binding to wild-type and T194A mutant of PrPC could be observed when these proteins were expressed in mammalian cells. As the T194A mutation has been proposed to alter the pattern of glycosylation, understanding the interaction in a more physiological context may be of value in predicting potential consequences of prion immunotherapy in animals bearing this mutation. Immunoprecipitation assays with lysates of wild-type or T194A bovine prion protein stably expressed in HEK293T cells () were conducted with antibodies SN6b, 6D11 and anti-FLAG (). Western blot analysis showed that SN6b immunoprecipitates bPrP(T194A) but not wild-type PrP () whereas 6D11 immunoprecipitated both wild-type bPrP and bPrP(T194A). No immunoprecipitation was observed in negative controls with anti-FLAG or beads alone.
Figure 7. The pathogenic mutant of bovine T194A which is a homolog of the T183A mutation in humans disrupts the glycosylation of PrPC and exposes an epitope that is only recognized in misfolded prion proteins. (A) Lysis followed by centrifugation of HEK293T cells stably expressing glycosylated wild-type bPrPC or T194A mutation (disrupted glycosylation) of bPrPC. (B) Immunoprecipitation of HEK293T cells stably expressing glycosylated wild-type PrPC or T194A mutation (disrupted glycosylation) of bPrPC with antibodies SN6b, 6D11 (positive control) and anti-FLAG (negative control). Bovine PrPC is not immunoprecipitated with antibody SN6b as compared with the T194A mutant of bPrPC that is immunoprecipitated, depicting a band having an intensity that is lower than that with antibody 6D11. The antibody 6D11 immunoprecipitated both wild-type or T194A mutant of bPrPC with almost similar band intensities for both proteins. The antibody anti-FLAG did not immunoprecipitate either wild-type or T194A mutant of bPrPC. The T194A mutant of bPrPC was not immunoprecipitated with antibody uncoupled beads as compared with for the E.coli expressed T194A of bPrPC. Molecular size markers in kDa are indicated on the left hand side of panel A.
Protease resistance
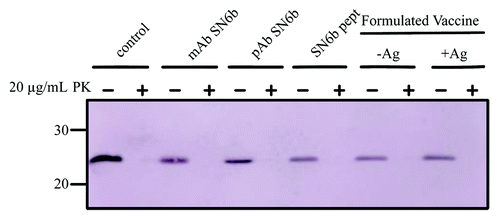
Antibodies targeted to the misfolded conformation of PrP have the theoretical potential to promote disease by serving as chaperones or templates that promote or stabilize the misfolded conformation. These concerns are minimized in situations where the antibody cannot bind to PrPC. As bPrP(T194A) exhibits greater structural propensity for misfolding, and we found that the SN6b antibody could bind to this mutant protein, we tested if the antibody could generate proteinase K (PK) resistant PrPSc from a bPrP(T194A) template. While nanopore analysis of bPrP(T194) under non-denaturing conditions, in the presence of the SN6b antibody, did not induce a detectable population of different conformation(s) (mostly type-II events) of PrP this does not rule out the possibility that PK resistant PrP could be formed as a consequence of prolonged incubation with the antibody alone (). To test this possibility the bPrP (T194A) recombinant protein was incubated with the SN6b antibody at 37 °C with shaking for 24 h to attempt formation of β-oligomeric PK-resistant recombinant PrP that can be induced under similar conditions but normally requires denaturant.Citation43 Formation of PK resistant PrPSc from bPrP(T194A) was assayed using a low concentration (20 µg/ml) of PK to sensitively detect if any PK resistant PrP was formed. No PK resistant protein could be detected when bPrP(T194A) was incubated with the SN6b antibody alone (). Formation of PK resistant PrPSc was also evaluated in the presence of the SN6b-Lkt fused immunizing peptide and formulated vaccine to determine if exposure to the antigenic peptide template might also be capable of inducing misfolding. We did not detect the formation of any PK resistant protein after incubating misfolding prone bPrP(T194A) with SN6b polyclonal or monoclonal antibody, the immunizing peptide or formulated vaccine ().
Figure 8. Antibodies and vaccines which are specific for the misfolded conformation of PrPC does not promote PK resistant PrPSc for E.coli expressed T194A mutant of bPrPC. In the absence of antibody, peptide or vaccine the T194A mutant of bPrPC is completely digested in PK. In the presence of mAb SN6b, pAb SN6b, peptide SN6b or formulated vaccines, -Ag (no antigen) and +Ag (with antigen) the T194A mutant of bPrPC is completely digested in PK. Molecular size markers in kDa are indicated on the left hand side of the panel.
Discussion
Previously our group optimized a YYR containing disease-specific epitope of the prion protein to develop a vaccine antigen that induces PrPSc-specific antibody responses.Citation20 This vaccine has considerable potential to serve as a tool for the control of animal prion diseases such as CWD that continues to spread, resulting in exposure of many animals to the infectious agent. In the case of human prion diseases administration of such a vaccine would be far more limited as exposure to infectious prions or generation of sporadic or familial forms of a prion are quite rare. The rates of prion disease in humans are not of sufficient magnitude to justify the vaccination of the general population. A human prion vaccine, if developed, would likely be utilized in a more strategic fashion, perhaps for prion researchers, individuals suspected of having consumed prion infected meat, individuals exposed to contaminated medical instruments or those with a genetic propensity for prion diseases.
There are a number of familial prion diseases that reflect different mutations within the prion protein gene.Citation27,Citation29 In these individuals immunization with a prion vaccine would seem a rationale course of action. We previously conducted experiments to confirm that antibodies produced in response to the YYR-based vaccine did not bind PrPC in “normal” individuals of a number of species. However, the theoretical consideration of administering the vaccine on individuals genetically predisposed to prion diseases prompted the question of whether the apparently PrPSc-specific antibodies would interact with a prion protein that is prone to misfold based on changes to its primary structure.
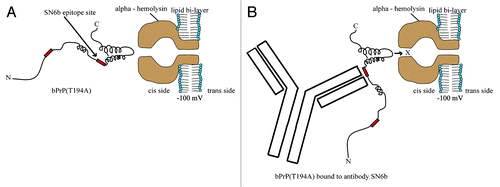
One rare genetic change in primary structure that has been identified to predispose individuals to prion disease is the human T183A mutation (corresponding to bovine T194A). To address the possibility that polyclonal antibodies specific for PrPSc might also bind a protein with the propensity to misfold, we used nanopore analysis, a sensitive single-molecule technique. Nanopore analysis of wild-type bPrP vs. bPrP(T194A) showed different values for blockade current (I) and time (T) due to interactions with the α-hemolysin pore. In the absence of a denaturant such as Gdn-HCl, antibody prion interactions were detected between the PrPSc-specific polyclonal antibody SN6b and bPrP(T194A) but not with the wild-type bPrP. In the presence of 1M Gdn-HCl, antibody-prion interactions were observed with both the wild-type and T194A mutant proteins, but with greater binding to the mutant. With nanopore analysis, specificity of the SN6b antibody for the mutant (T194A) and not wild-type was confirmed by lack of significant binding to the anti-FLAG antibody negative control. The binding of SN6b to the T194A mutant of PrPC may be due to the exposure of the epitope containing amino acids VYYRP that is buried in wild-type PrPC. Flexibility is conferred by the mutation T→A at position 194 that disrupts two hydrogen bonds between the second β-sheet and second α-helix of the prion protein ().Citation44 This flexibility also increases the propensity of the molecule to thread into the α-hemolysin pore, resulting in more type-II events.Citation36,Citation40 Once the SN6b antibody binds the protein, the antibody/prion complex is too large to intercalate through the α-hemolysin pore ().Citation35 In the case of the bovine wild-type PrPC the epitope for SN6b is buried in the globular domain of the protein and will not be affected by the addition of antibody.Citation19,Citation20,Citation45 Previously it was shown that wild-type bPrP generated about 35%‒40% of type-II events.Citation35,Citation37 This decrease in the number of type-II events in PrPC as compared with T194A may be due to the orientation of the molecule that may only thread into the pore through the three negatively charged amino acids (DYEDRYYR) located on the helix α1.Citation37 The interaction of the T194A mutant of PrPC with SN6b by nanopore analysis was validated through ELISAs and immunopreciptation of the mutant but not the wild-type proteins expressed in bacteria or mammalian cells.
Figure 9. The bPrP(T194A)/antibody complex is too large to intercalate whereas, unbound molecules are unaffected with α-hemolysin. A model for single molecule analysis using an α-hemolysin pore, illustrates the orientation of T194A mutant of bPrPC and antibody/prion complex. (A) The mutation T→A at position 194 disrupts two hydrogen bonds between the second β-sheet and second α-helix of PrP.Citation31 Therefore, now the more flexible T194A interacts with the α-hemolysin pore, increasing the number of type-II events. (B) The mutation T→A at position 194 may expose the epitope that binds SN6b and the antibody/prion complex is too large to intercalate through the α-hemolysin pore.
It seems clear that mutant prion protein is being recognized appropriately by the PrPSc-specific antibody. This could suggest that the mutant prion protein, which is associated with spontaneous early onset dementia, has a greater propensity to occupy conformations that are structurally consistent with the disease-associated folding pattern. This may also reflect specific misfolding to this conformation or perhaps greater flexibility within the protein which allows the antibody to select isoforms presenting the disease associated epitope from within a large population of structural isomers.Citation31 The binding of these disease-associated isoforms may have therapeutic value in neutralizing these species. The therapeutic value of this interaction will need to be evaluated through immunization of transgenic animal models of these familial prion diseases.
Collectively, these results support the value of conformation specific immunotherapy for the prion diseases. As well the concept of a DSE epitope may require specific consideration of the molecular context in which they are being applied. This could have significance for the use of vaccines for prion, as well as other, protein misfolding diseases. There are a number of human diseases, such as Alzheimer, Parkinson, Huntington, and ALS that are linked to protein misfolding. Prion diseases were also included within this category but usually with the important distinction of their infectious nature. This distinction is beginning to be questioned as there is emerging evidence that a number of these other diseases also self-propagate, although not infectious in the sense of being transmissible between individuals.Citation5-Citation9 This paradigm shift on the mechanisms of progression of these diseases is significant for a number of reasons, including renewed optimism that a vaccine may be able to halt progression of these diseases.Citation46 Attributing these unique characteristics to the misfolded form of these proteins also brings the issue of specificity to the forefront. Similar to the treatment of cancer, the challenge is to effectively manage, or eliminate, the pathological condition while preserving the functions of the healthy counterparts. These challenges are especially relevant to immunotherapy. In classical infectious diseases there is usually a clear distinction between self and non-self. For protein misfolding disorders the line is less clear, self is represented by the properly folded conformation while the vaccine target is the misfolded dysfunctional species. This is not trivial as there are concerns of the pathological consequences of potential autoimmunity. Fortunately there is increasing evidence that conformation-specific immunotherapy is possible through the identification of DSEs which elicit potent immune responses highly specific to the misfolded conformation. Vaccines based on DSEs, have been investigated for a number of diseases including prion disease,Citation20 ALSCitation47,Citation48 Alzheimer, Parkinson, and Huntington diseases (reviewed in ref. Citation46). Investigations demonstrating that induced anti-DSE antibodies do not recognize correctly folded protein are paramount and must take into account the variance of immune response within a genetically diverse population which includes individuals who are genetically disposed to the disease.
Materials and Methods
Recombinant proteins, antibodies and α-hemolysin pore
Escherichia coli (E.coli) expressed wild-type bovine prion (bPrP) (positions 25 [start sequence, KKRPK] to 242 [stop sequence RGAS]) and bovine prion protein with mutation T194A (bPrP[T194A]) (positions 25 to 242) were purchased from (PR-370 and PR-371; Jena Bioscience) and dissolved in 10 mM TRIS-HCl, 0.1 mM EDTA pH 8.0 at 1 mg/ml for non-denaturing conditions. The 1 mg/ml protein solution was pre-incubated with 1 M Gdn-HCl for 1 h to allow partial unfolding. For nanopore experiments, the whole aliquot was then added to the cis side of the cup, proximal to the aperture. We also used wild-type bovine prion containing amino acid residues (25‒242) expressed and purified as described previously.Citation49 Monoclonal antibody (mAb) M2188 (IgG2b) which recognizes amino acids 146 to 153 (SRPLIHFG) in PrPC was generated as described in Andrievskaia et al.Citation49 Polyclonal antibody (pAb) SN6b, was raised against a highly immunogenic presentation of the following epitope QVYYRPVDQYSNQN in the form (forward-back-back)4 in sheep and affinity purified (Covance) from sheep serum.Citation20 Polyclonal antibodies were selected to mimic the situation of vaccination and detect binding to all conformations explored by the mutant T194A of PrPC. A mouse monoclonal antibody raised against the cervid form of the SN6b epitope QVYYRPVDQYNNQN (Immunoprecise Antibodies Ltd.) was also tested in the in vitro protein misfolding assay. Monoclonal antibodies, 6D11 (recognizes epitope 93–109 in PrPC; that binds both PrPC and PrPSc) and anti-FLAG (binds FLAG peptide: DYKDDDDK) were purchased from (PrP-6D11:sc-58581; Santa Cruz Biotechnology, Inc. and F1804; Sigma-Aldrich) respectively. α-Hemolysin was purchased from (H9395; Sigma-Aldrich) and made at 2 μg/ml in 1.0 M KCl in 10 mM TRIS-HCl buffer at pH 7.8.
Nanopore analysis
Nanopore analysis was employed to investigate the binding of E. coli expressed recombinant wild-type bovine prion protein (bPrP) and prion protein with mutation T194A (bPrP[T194A]), by M2188 which binds the epitope SRPLIHFG in bPrP and bPrP(T194A) as well as SN6b, that is specific for the misfolded conformation.
As previously described in detail, nanopore analysis was conducted using an Axopatch 200B amplifier (Axon Instruments) connected to a CV203BU head-stage (Axon Instruments) at 22 ± 1 °C with an applied potential of −100 mV at a bandwidth of 10 KHz.Citation33-Citation35 After insertion of an α-hemolysin pore, 20 µl of protein solution (1 mg/ml) was added to the 1.5 ml cis chamber proximal to the aperture.Citation34,Citation36 For some experiments the protein was partially unfolded (or conformationally-relaxed) by incubating with 1M Gdn-HCl for 15 min at 22 °C. The protein was then incubated with antibody for 1 h before addition of the complete sample to the nanopore chamber. The electrolyte solution for nanopore experiments was 1.0 M KCl in 10 mM TRIS-HCl buffer at pH 7.8 with a maximum Gdn-HCl concentration of 20 mM. The populations for blockade current were obtained by fitting the blockade current distribution with the Gaussian function. The lifetime data was acquired by fitting each blockade duration distribution with a single exponential function.Citation34,Citation35
Immunoprecipitation of recombinant prion protein
Tosyl-activated M-280 Dynabeads, were purchased from Invitrogen (14203; Life Technologies). The antibody (ligand) coupling was performed according to manufacturer’s instructions. Immunoprecipitation was performed using 8 µM recombinant bPrP or bPrP(T194A) that was diluted 1/18 with PBS containing 1 mg/ml of BSA to a final volume of 450 µl. Dynabeads (48 µl) coupled to 20 µg of antibody were added to this protein solution and incubated on a nutator at 37 °C for 1–3 h. Then the beads were collected on a magnetic stand, washed 5 times with 1 ml RIPA buffer (20 mM Tris, 300 mM NaCl, 2% w/v deoxycholic acid, 2% w/v Igepal, pH 7.5) and finally washed with 200 µl low salt buffer (20 mM TRIS-HCl, pH 7.5). The proteins were eluted with 50 µl of 2 × Laemmli SDS-PAGE loading dye containing 0.5% v/v β-mercaptoethanol and boiled for 5 min at 95 °C. The samples were then cooled to room temperature and centrifuged at 13 000 rpm for 1 min prior to analysis by 12% polyacrylamide SDS-PAGE and western blotting to detect recombinant prion protein. Proteins separated by SDS-PAGE were transferred in Tris-Glycine transfer buffer, pH 8.3 at 140 mA overnight. The nitrocellulose blots were blocked for 1 h at room temperature with 5% non-fat milk in TBS-T (10 mM TRIS-HCl, pH 7.5, 150 mM NaCl, 0.05% Tween-20) and detected using primary antibody 6D11 in 5% skim Milk TBS-T and secondary antibody Clean-Blot™ IP detection reagent (P121233; Thermo-Fisher) in 5% skim Milk in TBS-T.
Cloning and eukaryotic expression of wild-type or T194A bovine prion protein
The bovine prion ORF was PCR amplified from genomic DNA isolated from lung tissue with primers 5′-GAACGCTAGC CACCATGGTG AAAAGCCACA TAGG-3′and 5′-GAGGAATTCG GTTGCCCCTA TCCTACTATG AG-3′. The ORF was proceeded by a Kozak sequence and cloned downstream of a human CMV promoter contained within an episomal vector containing the EBNA-1 antigen ORF and P origin. A puromycin-resistant gene allowed for the selection and stable maintenance of the vector. The T194A mutation was created by inverse PCR with primers 5′-GCCGTCAAGG AACACACAGT CAC-3′ and 5′-GATATTGACA CAGTCATGCA CAAAG-3′ using as template a plasmid containing the wild-type bovine prion ORF cloned as above. Following mutagenesis, the ORF was sub-cloned into the episomal vector. All sequences were verified by DNA sequencing and confirmed to be correct. Wild-type bovine prion constructs encoded for protein was identical to the National Centre for Biotechnology Information, accession number; NP_851358.2).
Stable expression of wild-type or T194A mutant PrPC in cell culture
Human embryonic kidney (HEK293T) cells were transfected with plasmids containing the wild-type or T194A mutant of PrPC using Turbofect transfectant reagent (R0531; Thermo Scientific) according to manufacturer’s instructions. Transfected cells were grown in Dulbecco’s Modified Eagle Medium (DMEM) from (D5796; Sigma-Aldrich) containing 10% fetal bovine serum (FBS) with puromycin (1 µg/ml) to select for the episomal vector. Cultures were incubated with 10% CO2, at 37 °C.
Immunoprecipitation of HEK293T cells stably expressing wild-type or T194A PrPC
Cells were grown to confluency, detached and washed with cold Dulbecco’s PBS followed by centrifugation at 311 × g for 5 min at 4 °C. Cells were then lysed in NP-40 buffer (150 mM NaCl, 50 mM HEPES, 1% NP-40, pH 7.4) supplemented with protease inhibitors (R1321; ProteoBlock, Fermentas, Thermo Scientific) for 30 min at 4 °C with gentle shaking followed by centrifugation at 10 000 rpm for 10 min at 4 °C to remove cell debris. Each reaction for immunoprecipitation was performed in a 500 µl volume. Antibody was added (SN6b, 6D11 [positive control], anti-FLAG [negative control]) to cell lysate and incubated at 4 °C on a nutator for 16 h: this was followed by the addition of 50‒100 µl of µMACS protein G micro beads (130–071–101; Miltenyi Biotec) and incubation for 3 h at 4 °C on a nutator. A µMAC column (130–042–701; Miltenyi Biotec) was placed on a magnetic stand and washed with 200 µl of NP-40 buffer. The lysate was then added to the column matrix and the non-bound flow-through was collected and analyzed for protein. The column was washed 4 times with 200 µl of NP-40 buffer and finally with 100 µl of low salt buffer (20 mM TRIS-HCl, pH 7.5). For protein elution, first 20 µl of pre-heated (95 °C) 1 × SDS gel loading buffer (50 mM Tris- HCl, 50 mM DTT. 1% SDS, 0.005% bromophenol blue, 10% glycerol) was added and incubated at room temperature for 5 min. This flow through was removed, and a fresh collection tube was placed at the tip of the column. Protein was eluted with 50 µl pre-heated (95 °C) 1 × SDS gel loading buffer and was analyzed by SDS-PAGE. The samples were analyzed by 12% polyacrylamide SDS-PAGE and western blotting to detect prion protein. Proteins separated by SDS-PAGE were transferred in Tris-Glycine transfer buffer at 250 mA for 1.5 h. The nitrocellulose blots were blocked for 1 h at room temperature with 5% non-fat milk in TBS-T and detected using primary antibody 6D11 (dilution 1:200) in 5% skim Milk TBS-T and secondary antibody Clean-Blot™ (dilution1:100) IP detection reagent.
Protease resistance
Misfolding experiments were performed in a total volume of 40 µl of sterile PBSA by combining 2 µg of E. coli expressed recombinant protein with an equivalent quantity of monoclonal antibody, polyclonal antibody or SN6b-Leukotoxin fusion peptide (in 1‒3 µl). To test formulated vaccines, 1 µl of each was added to the antibody protein mix (1 µl of PBS was included as control) and reactions were incubated for 24 h at 37 °C with shaking at 200 rpm. After brief centrifugation to bring down liquid, 20 µl was digested with 20 µg/ml of Proteinase K (PK) (P-6556; Sigma-Aldrich) at 37 °C for 30 min with shaking. Undigested controls were incubated the same way with an equivalent volume of PBS. Digests were halted by adding PMSF to a final concentration of 2 mM. Samples were combined with an equal volume of 2 × Laemmli SDS-PAGE loading dye containing 0.5% v/v β-mercaptoethanol, prior to analysis by 12% polyacrylamide SDS-PAGE and western blotting.
ELISA
Antibody binding was quantified by ELISA. The 96-well polystyrene microtiter plates (Immulon 2HB, Dynatech) were coated overnight with 0.5 μg per well of bovine bPrP(T194A) or wild-type bovine from Jena Bioscience. Plates were washed 5 times with distilled water (dH2O) then blocked with 200 μl per well of TBS-T and 1% skim milk. Plates were incubated for 1 h at room temperature then washed 5 times with dH2O. Antibodies SN6b, M2188 and anti-FLAG were diluted 1:40 in TBS-T with 1% skim milk and then serially diluted 1:4 in TBS-T plus 1% skim milk. Plates were incubated for 1 h, washed 5 times with dH2O, then 100 μl of alkaline phosphatase (AP)-conjugated rabbit anti-sheep immunoglobulin gamma (IgG) or AP-conjugated goat anti-mouse IgG (Kirkgaard and Perry Laboratories; 1:4000) were added to each well to detect bound ovine and murine antibody, respectively. After the 1 h incubation, plates were washed 5 times with dH2O then 100 μl p-nitrophenyl phosphate (PNPP) (Sigma) was added to each well for 1 h. ELISA titers were expressed as the reciprocal of the highest serum dilution resulting in a reading exceeding two standard deviations above the negative control.
| Abbreviations: | ||
| PrPC | = | cellular prion protein |
| PrPSc | = | scrapie prion protein |
Acknowledgments
This work was supported by PrioNet, Saskatchewan Agriculture and Development and Alberta Prion Research Institute.
Submitted
05/01/2013
Revised
05/17/2013
Accepted
05/22/2013
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
References
- Hedlin P, Taschuk R, Potter A, Griebel P, Napper S. Detection and Control of Prion Diseases in Food Animals. ISRN Vet Sci 2012; 2012:1 - 24; http://dx.doi.org/10.5402/2012/254739
- Beekes M. Prions and prion diseases. FEBS J 2007; 274:575; http://dx.doi.org/10.1111/j.1742-4658.2006.05629.x; PMID: 17288546
- Collinge J. Prion diseases of humans and animals: their causes and molecular basis. Annu Rev Neurosci 2001; 24:519 - 50; http://dx.doi.org/10.1146/annurev.neuro.24.1.519; PMID: 11283320
- Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science 1982; 216:136 - 44; http://dx.doi.org/10.1126/science.6801762; PMID: 6801762
- Prusiner SB. Cell biology. A unifying role for prions in neurodegenerative diseases. Science 2012; 336:1511 - 3; http://dx.doi.org/10.1126/science.1222951; PMID: 22723400
- Grad LI, Guest WC, Yanai A, Pokrishevsky E, O’Neill MA, Gibbs E, et al. Intermolecular transmission of superoxide dismutase 1 misfolding in living cells. Proc Natl Acad Sci U S A 2011; 108:16398 - 403; http://dx.doi.org/10.1073/pnas.1102645108; PMID: 21930926
- Luk KC, Kehm V, Carroll J, Zhang B, O’Brien P, Trojanowski JQ, et al. Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 2012; 338:949 - 53; http://dx.doi.org/10.1126/science.1227157; PMID: 23161999
- Miller G. Neurodegeneration. Could they all be prion diseases?. Science 2009; 326:1337 - 9; http://dx.doi.org/10.1126/science.326.5958.1337; PMID: 19965731
- Polymenidou M, Cleveland DW. Prion-like spread of protein aggregates in neurodegeneration. J Exp Med 2012; 209:889 - 93; http://dx.doi.org/10.1084/jem.20120741; PMID: 22566400
- Baron T, Biacabe AG. Origin of bovine spongiform encephalopathy. Lancet 2006; 367:297 - 8, author reply 298-9; http://dx.doi.org/10.1016/S0140-6736(06)68060-4; PMID: 16443028
- Biacabe AG, Morignat E, Vulin J, Calavas D, Baron TG. Atypical bovine spongiform encephalopathies, France, 2001-2007. Emerg Infect Dis 2008; 14:298 - 300; http://dx.doi.org/10.3201/eid1402.071141; PMID: 18258124
- Williams ES. Chronic wasting disease. Vet Pathol 2005; 42:530 - 49; http://dx.doi.org/10.1354/vp.42-5-530; PMID: 16145200
- Sigurdson CJ, Aguzzi A. Chronic wasting disease. Biochim Biophys Acta 2007; 1772:610 - 8; http://dx.doi.org/10.1016/j.bbadis.2006.10.010; PMID: 17223321
- Li L, Napper S, Cashman NR. Immunotherapy for prion diseases: opportunities and obstacles. Immunotherapy 2010; 2:269 - 82; http://dx.doi.org/10.2217/imt.10.3; PMID: 20635933
- Mouillet-Richard S, Ermonval M, Chebassier C, Laplanche JL, Lehmann S, Launay JM, et al. Signal transduction through prion protein. Science 2000; 289:1925 - 8; http://dx.doi.org/10.1126/science.289.5486.1925; PMID: 10988071
- Solforosi L, Criado JR, McGavern DB, Wirz S, Sánchez-Alavez M, Sugama S, et al. Cross-linking cellular prion protein triggers neuronal apoptosis in vivo. Science 2004; 303:1514 - 6; http://dx.doi.org/10.1126/science.1094273; PMID: 14752167
- Arsenault RJ, Li Y, Potter A, Griebel PJ, Kusalik A, Napper S. Induction of ligand-specific PrP (C) signaling in human neuronal cells. Prion 2012; 6:477 - 88; http://dx.doi.org/10.4161/pri.21914; PMID: 22918447
- Roettger Y, Du Y, Bacher M, Zerr I, Dodel R, Bach JP. Immunotherapy in prion disease. Nat Rev Neurol 2013; 9:98 - 105; http://dx.doi.org/10.1038/nrneurol.2012.258; PMID: 23247613
- Paramithiotis E, Pinard M, Lawton T, LaBoissiere S, Leathers VL, Zou WQ, et al. A prion protein epitope selective for the pathologically misfolded conformation. Nat Med 2003; 9:893 - 9; http://dx.doi.org/10.1038/nm883; PMID: 12778138
- Hedlin PD, Cashman NR, Li L, Gupta J, Babiuk LA, Potter AA, et al. Design and delivery of a cryptic PrP(C) epitope for induction of PrP(Sc)-specific antibody responses. Vaccine 2010; 28:981 - 8; http://dx.doi.org/10.1016/j.vaccine.2009.10.134; PMID: 19925901
- Coulthart MB, Cashman NR. Variant Creutzfeldt-Jakob disease: a summary of current scientific knowledge in relation to public health. CMAJ 2001; 165:51 - 8; PMID: 11468957
- Aguzzi A. Prion diseases of humans and farm animals: epidemiology, genetics, and pathogenesis. J Neurochem 2006; 97:1726 - 39; http://dx.doi.org/10.1111/j.1471-4159.2006.03909.x; PMID: 16805779
- Hsiao K, Prusiner SB. Inherited human prion diseases. Neurology 1990; 40:1820 - 7; http://dx.doi.org/10.1212/WNL.40.12.1820; PMID: 2247227
- Windl O, Dempster M, Estibeiro JP, Lathe R, de Silva R, Esmonde T, et al. Genetic basis of Creutzfeldt-Jakob disease in the United Kingdom: a systematic analysis of predisposing mutations and allelic variation in the PRNP gene. Hum Genet 1996; 98:259 - 64; http://dx.doi.org/10.1007/s004390050204; PMID: 8707291
- Hsiao K, Baker HF, Crow TJ, Poulter M, Owen F, Terwilliger JD, et al. Linkage of a prion protein missense variant to Gerstmann-Sträussler syndrome. Nature 1989; 338:342 - 5; http://dx.doi.org/10.1038/338342a0; PMID: 2564168
- Glatzel M, Ott PM, Linder T, Gebbers JO, Gmür A, Wüst W, et al. Human prion diseases: epidemiology and integrated risk assessment. Lancet Neurol 2003; 2:757 - 63; http://dx.doi.org/10.1016/S1474-4422(03)00588-X; PMID: 14636781
- Kovács GG, Trabattoni G, Hainfellner JA, Ironside JW, Knight RS, Budka H. Mutations of the prion protein gene phenotypic spectrum. J Neurol 2002; 249:1567 - 82; http://dx.doi.org/10.1007/s00415-002-0896-9; PMID: 12420099
- Nitrini R, Rosemberg S, Passos-Bueno MR, da Silva LS, Iughetti P, Papadopoulos M, et al. Familial spongiform encephalopathy associated with a novel prion protein gene mutation. Ann Neurol 1997; 42:138 - 46; http://dx.doi.org/10.1002/ana.410420203; PMID: 9266722
- Finckh U, Müller-Thomsen T, Mann U, Eggers C, Marksteiner J, Meins W, et al. High prevalence of pathogenic mutations in patients with early-onset dementia detected by sequence analyses of four different genes. Am J Hum Genet 2000; 66:110 - 7; http://dx.doi.org/10.1086/302702; PMID: 10631141
- Kiachopoulos S, Bracher A, Winklhofer KF, Tatzelt J. Pathogenic mutations located in the hydrophobic core of the prion protein interfere with folding and attachment of the glycosylphosphatidylinositol anchor. J Biol Chem 2005; 280:9320 - 9; http://dx.doi.org/10.1074/jbc.M412525200; PMID: 15591591
- van der Kamp MW, Daggett V. Pathogenic mutations in the hydrophobic core of the human prion protein can promote structural instability and misfolding. J Mol Biol 2010; 404:732 - 48; http://dx.doi.org/10.1016/j.jmb.2010.09.060; PMID: 20932979
- Chebaro Y, Derreumaux P. The conversion of helix H2 to beta-sheet is accelerated in the monomer and dimer of the prion protein upon T183A mutation. J Phys Chem B 2009; 113:6942 - 8; http://dx.doi.org/10.1021/jp900334s; PMID: 19371053
- Sutherland TC, Long YT, Stefureac RI, Bediako-Amoa I, Kraatz HB, Lee JS. Structure of peptides investigated by nanopore analysis. Nano Lett 2004; 4:1273 - 7; http://dx.doi.org/10.1021/nl049413e
- Stefureac R, Long YT, Kraatz HB, Howard P, Lee JS. Transport of alpha-helical peptides through alpha-hemolysin and aerolysin pores. Biochemistry 2006; 45:9172 - 9; http://dx.doi.org/10.1021/bi0604835; PMID: 16866363
- Madampage CA, Andrievskaia O, Lee JS. Nanopore detection of antibody prion interactions. Anal Biochem 2010; 396:36 - 41; http://dx.doi.org/10.1016/j.ab.2009.08.028; PMID: 19699704
- Meng H, Detillieux D, Baran C, Krasniqi B, Christensen C, Madampage C, et al. Nanopore analysis of tethered peptides. J Pept Sci 2010; 16:701 - 8; http://dx.doi.org/10.1002/psc.1289; PMID: 20814890
- Stefureac RI, Madampage CA, Andrievskaia O, Lee JS. Nanopore analysis of the interaction of metal ions with prion proteins and peptides. Biochem Cell Biol 2010; 88:347 - 58; http://dx.doi.org/10.1139/O09-176; PMID: 20453935
- Madampage CA, Tavassoly O, Christensen C, Kumari M, Lee JS. Nanopore analysis: An emerging technique for studying the folding and misfolding of proteins. Prion 2012; 6:116 - 23; http://dx.doi.org/10.4161/pri.18665; PMID: 22421211
- Oukhaled G, Mathé J, Biance AL, Bacri L, Betton JM, Lairez D, et al. Unfolding of proteins and long transient conformations detected by single nanopore recording. Phys Rev Lett 2007; 98:158101 - 4; http://dx.doi.org/10.1103/PhysRevLett.98.158101; PMID: 17501386
- Stefureac R, Waldner L, Howard P, Lee JS. Nanopore analysis of a small 86-residue protein. Small 2008; 4:59 - 63; http://dx.doi.org/10.1002/smll.200700402; PMID: 18058890
- Baran C, Smith GS, Bamm VV, Harauz G, Lee JS. Divalent cations induce a compaction of intrinsically disordered myelin basic protein. Biochem Biophys Res Commun 2010; 391:224 - 9; http://dx.doi.org/10.1016/j.bbrc.2009.11.036; PMID: 19903451
- Tavassoly O, Lee JS. Methamphetamine binds to α-synuclein and causes a conformational change which can be detected by nanopore analysis. FEBS Lett 2012; 586:3222 - 8; http://dx.doi.org/10.1016/j.febslet.2012.06.040; PMID: 22771474
- Baskakov IV, Legname G, Baldwin MA, Prusiner SB, Cohen FE. Pathway complexity of prion protein assembly into amyloid. J Biol Chem 2002; 277:21140 - 8; http://dx.doi.org/10.1074/jbc.M111402200; PMID: 11912192
- Riek R, Wider G, Billeter M, Hornemann S, Glockshuber R, Wüthrich K. Prion protein NMR structure and familial human spongiform encephalopathies. Proc Natl Acad Sci U S A 1998; 95:11667 - 72; http://dx.doi.org/10.1073/pnas.95.20.11667; PMID: 9751723
- López Garcia F, Zahn R, Riek R, Wüthrich K. NMR structure of the bovine prion protein. Proc Natl Acad Sci U S A 2000; 97:8334 - 9; http://dx.doi.org/10.1073/pnas.97.15.8334; PMID: 10899999
- Southwell AL, Patterson PH. Antibody therapy in neurodegenerative disease. Rev Neurosci 2010; 21:273 - 87; http://dx.doi.org/10.1515/REVNEURO.2010.21.4.273; PMID: 21086760
- Rakhit R, Robertson J, Vande Velde C, Horne P, Ruth DM, Griffin J, et al. An immunological epitope selective for pathological monomer-misfolded SOD1 in ALS. Nat Med 2007; 13:754 - 9; http://dx.doi.org/10.1038/nm1559; PMID: 17486090
- Liu HN, Tjostheim S, Dasilva K, Taylor D, Zhao B, Rakhit R, et al. Targeting of monomer/misfolded SOD1 as a therapeutic strategy for amyotrophic lateral sclerosis. J Neurosci 2012; 32:8791 - 9; http://dx.doi.org/10.1523/JNEUROSCI.5053-11.2012; PMID: 22745481
- Andrievskaia O, McRae H, Elmgren C, Huang H, Balachandran A, Nielsen K. Generation of antibodies against bovine recombinant prion protein in various strains of mice. Clin Vaccine Immunol 2006; 13:98 - 105; http://dx.doi.org/10.1128/CVI.13.1.98-105.2006; PMID: 16426006