Abstract
The prion protein (PrP), a GPI-anchored glycoprotein, is inefficiently secreted by mammalian microsomes, 50% being found as transmembrane (TM) proteins with the central TM1 segment spanning the membrane. TM1 hydrophobicity is marginal for lateral membrane insertion, which is primarily driven by hydrophobic interaction between the ER translocon and substrates in transit. Most inserted TM1 has its N-terminus in the ER lumen (Ntm orientation), as expected for arrest of normal secretion. However, 20% is found in inverted Ctm orientation. These are minor species in vivo, presumably a consequence of efficient quality control. PrP mutations that increase TM1 hydrophobicity result in increased Ctm insertion, both in vitro and in mouse brain, and a strong correlation is found between CtmPrP insertion and neuropathology in transgenic mice; a copper-dependent pathogenicity mechanism is suggested. PrP fusions with a C-terminal epitope tag, when expressed in yeast cells at moderate levels, appear to interact efficiently with the translocon, providing a useful model for testing the effects of PrP mutations on TM insertion and orientation. However, secretion of PrP by the mammalian translocon requires the TRAP complex, absent in yeast, where essentially all PrP ends up as TM species, 85–90% Ntm and 10–15% Ctm. Although yeast is, therefore, an incomplete mimic of mammalian PrP trafficking, effects on Ctm insertion of mutations increasing TM1 hydrophobicity closely reflect those seen in vitro. Electrostatic substrate-translocon interactions are a major determinant of TM protein insertion orientation and the yeast model was used to investigate the role of the large negative charge difference across TM1, a likely cause of translocation delay that would favor TM insertion and Ctm orientation. An increase in ΔCh from −5 to −7 caused a marked increase in Ctm insertion, while a decrease to −3 or −1 allowed 35 and about 65% secretion, respectively. Utility of the yeast model and the role of this charge difference in driving PrP membrane insertion are confirmed.
Introduction
The infectious agents in transmissible spongiform encephalopathies (TSE’s), untreatable and invariably fatal neurodegenerative diseases, are oligomeric misfolded forms of prion proteins (PrP’s) named PrPSc after Scrapie, the archetypal TSE of sheep.Citation1 PrP is expressed in many tissues, most strongly in neurons and lymphoid tissues. Familial prion diseases all result from mutations in the Prnp PrP gene, but not all are transmissible. Conversely, PrPSc is not directly toxicCitation2; interaction with cells that are actively producing normal PrP results in autocatalytic conversion of PrP to PrPSc and its accumulation.Citation2 This, in turn, apparently causes production of misfolded or mislocalized neurotoxic forms of PrP.Citation3 The unique susceptibility of neurons is presumably related to their longevity and the cumulative effects of direct toxicity of the misfolded protein or of responses to the stress of this burden.Citation4 Neurotoxicity in transgenic mouse models of certain pathogenic Prnp mutations, such as the A117V mutation that causes Gerstmann-Sträussler-Scheinker syndrome and other mutations that increase TM1 hydrophobicity, correlates strongly with increased production of a form of PrP in which the central hydrophobic TM1 segment (residues 112–134; ) spans the membrane in Ctm orientation, with the C-terminus in the ER lumen.Citation5 Using yeast as a model system for PrP expression, we now demonstrate that these PrP mutations have similar effects on Ctm TM insertion in yeast cells and that a major risk factor for CtmPrP production is the large negative charge difference across TM1.
Figure 1. PrP sequence emphasizing segments relevant to this paper. The sequence elements shown are nearly identical in Hu, SHa and MH2M PrP’s, sharing all of the charged residues shown. MH2M, a mouse-hamster chimera, lacks Gly55. Hu numbering is used. The TM1 segment (112–134) and terminal signal sequences are shown as gray boxes. The mutations shown include KH-II (K110I, H111I) and A3V (A113V, A115V, A117V), principally affecting TM1 hydrophobicity, and P102L, all studied in mice.Citation37 The R136D, K106E and K110E mutations alter ΔCh across TM1 and the TSE25 (K23T, K24S, R25E) mutation at the mature N-terminus alters ΔCh across the signal peptide.
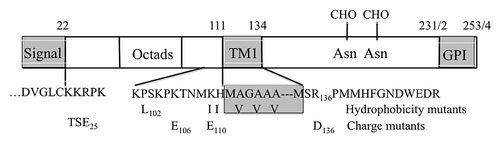
Although PrP’s are non-essential in mice, their sequences are highly conserved in mammals. The human and Syrian hamster (SHa) PrP precursors have 253 and 254 residues while MuPrP has 254 but lacks Gly55. Human numbering is used below. Twenty-two residue N-terminal secretion and C-terminal GPI anchor attachment signals are removed during translocation of the mature sequence (residues 23–231/2; ). This is expressed on cell surfaces in the normal cellular form PrPC, a GPI-anchored secreted protein of uncertain function that has two C-terminal sites for N-glycosylation. TM1 is partly exposed on the surface of the globular C-terminal domain in PrPC while the preceding N-terminal segment has no discernable structure in solution.Citation6 This segment contains 5 octad repeats that are able to bind copper or zinc ions and PrP endocytosis may play a role in governing cellular content of these ions.Citation7,Citation8 All of these features are conserved, so are apparently under strong selection.
In order for PrP to reach its normal GPI-linked cell surface location, it needs to be secreted. Its signal peptide must be recognized by the signal recognition particle (SRP) as it exits the ribosome so that it can be guided by the SRP receptor to the Sec61p ER translocon channel. Here it needs to enter in Ctm orientation to open the channel and initiate translocation of the mature sequence whose N-terminus, after signal peptidase cleavage, will have Ntm orientation in the ER lumen. Following translocation of the TM1 segment, core glycosylation occurs at one or both of the C-terminal acceptor sites. Arrival of the GPI anchor signal initiates the final maturation of the secreted PrPC peptide whose carbohydrate chains mature during Golgi transit. The conserved PrP sequence, however, seems far from optimal to achieve these results. Most significant are the presence of a sub-optimal secretion signal and the presence of the hydrophobic TM1 segment, long enough for TM insertion but of marginal hydrophobicity.Citation9,Citation10 In addition, TM1 is flanked by multiple charged residues (). These 11 and 14 residue flanking segments provide a net charge difference ΔCh (C minus N) across TM1 of about −5. Because of this large negative ΔCh, the preferred orientation of TM1 insertion into the translocon channel would be Ctm rather than the Ntm orientation required for continued secretion.Citation11-Citation13 This disparity may cause a delay in PrP translocation, promoting lateral insertion of TM1 into the ER membrane. However, unless the already translocated N-terminus is returned to the cytoplasm, insertion would necessarily be Ntm.
PrP is co-translationally secreted by mammalian microsomes, however efficiency is only about 50% and TM forms in both Ntm (40%) and Ctm (10%) orientations, anchored by the TM1 segment, are also produced.Citation5 Production of TM forms of the normal PrP sequence is difficult to detect in mouse brain, probably due to greater in vivo translocation efficiency and the efficacy of normal quality control and turnover mechanisms. Improved techniques, however, have allowed their detectionCitation14 and Ctm insertion becomes readily detectable in transgenic mice expressing PrP mutants that increase TM1 hydrophobicity.Citation5 CtmPrP produced in vivo was shown to be fully glycosylated and GPI anchored, with a mature N-terminus.Citation14 Fully cytoplasmic CyPrP, a minor species also observed in vivo,Citation14 may also be toxic since both are increased by pathogenic PrP mutations. The TM forms accumulate in a Golgi-like post-ER compartment.Citation4 It is hypothesized that PrPSc-induced enhanced accumulation of either CtmPrP or CyPrP is the proximal cause of neurotoxicity in TSE’s.Citation3 NtmPrP has no apparent role in pathogenicity.
We have employed yeast (Saccharomyces cereviseae), a model eukaryote unparalleled in the ease with which its genome and physiology can be manipulated, to investigate the role of charged residues in determining TM protein topology during ER insertion.Citation12,Citation15,Citation16 Yeast cells should be well suited for investigation of PrP secretion and TM insertion. However, while the basic machinery of protein translocation is conserved in all eukaryotes, complexities have evolved and it was first necessary to evaluate the fidelity with which the yeast secretion machinery would respond to mutations that increase TM1 hydrophobicity. In this paper we demonstrate that, when PrP fusion species are expressed at moderate levels in yeast, interaction with and gating of the Sec61 translocon channel appears to be quite efficient. The response of TM insertion orientation to TM1 hydrophobicity does reflect that seen in mammalian cells, however secretion is minimal and TM1 is almost completely trapped as a TM domain. If TM1 ΔCh is markedly reduced, however, secretion in yeast becomes quite efficient, demonstrating the propensity of TM1 ΔCh for controlling PrP translocation and implying the need for suppression of this effect in mammalian cells. Yeast cells, therefore, provide a tractable model for testing the effects of PrP sequence on translocation efficiency and TM insertion orientation.
Results
PrP constructs for yeast expression
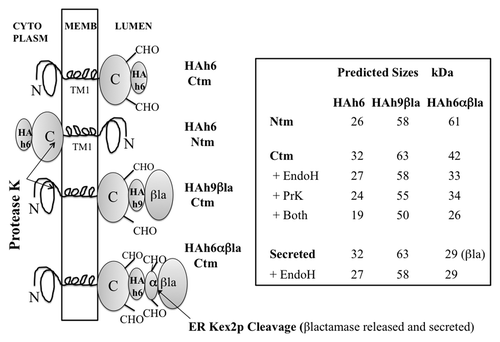
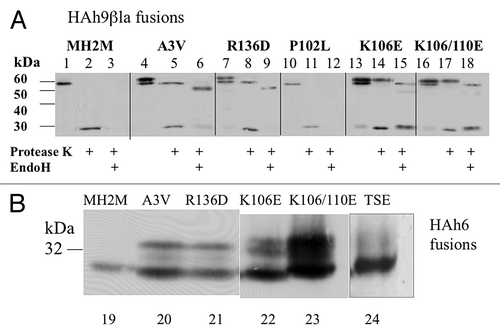
The vector pPGKT7g, derived from pS79-PB,Citation12 allows efficient translation of T7 RNA polymerase run-off transcripts in the rabbit reticulocyte system, and constitutive expression in yeast cells from the PGK promoter, but only at about 20% of the level provided by the parent pS79-PB vector (not shown). Reading frames comprising the Syrian hamster (SHa) and mouse/hamster chimera (MH2M) PrP’s and their KH-II and A3V mutantsCitation5 (, legend) were cloned into pPGKT7g from plasmids kindly provided by Dr Lingappa. The C-terminal GPI anchors, presumably irrelevant for early steps in ER translocation, were replaced by the triple hemagglutinin (HA) epitope tag and a His6 C-terminal sequence, producing PrP-HAh6 fusions (). HA tagged species in 1–10 μg of total protein extracts afforded robust western blot signals using anti-HA monoclonal antisera. C-terminal addition of the αβla reporterCitation12 produced PrP-HAh6-αβla fusions (), in which β−lactamase secretion acts as an independent reporter of C-terminal translocation. PrP-HAh9-βla fusions () lack the α peptide and remain intact when their C-termini are translocated, allowing the use of monoclonal anti-βlactamase sera for topology analysis by western blot. Only Ctm and secreted species are glycosylated; the predicted sizes of all products, after signal peptidase cleavage, are listed in .
Figure 2. PrP fusions expressed in yeast. Left; models of PrP fusions spanning the membrane. From the top, an HAh6 Ctm fusion, an HAh6 Ntm fusion, an HAh9βla Ctm fusion and an HAh6αβla Ctm fusion. Ntm forms of the latter species would also be inverted about the TM1 segment. The unstructured mature N-terminal segment is shown as a loop, TM1 as a four-loop helix and the globular C-terminal segment as an ovoid. In both Ctm and secreted forms this segment may be glycosylated at two sites, indicated by CHO. In Ntm orientation the C-terminal segments are retained in the cytoplasm and not glycosylated. HA is the triple HA tag, h6 and h9 are His6 and His9 Ni-binding tags, βla is the mature sequence of β-lactamase, as normally secreted, and α is a 30 residue fragment of pre-pro-α factor that includes two additional N-glycosylation sites and a site for cleavage by ER-Kex2p. Cleavage releases βlactamase in the ER lumen for secretion. Right; predicted sizes in kDa are shown, assuming that, in Ctm and secreted forms, the secretion signal is removed and all sites receive core N-glycosylation. Protease K treatment of intact microsomes should remove the HA tags from Ntm forms, should cleave about 8 kDa from the N-terminus of Ctm forms but should not affect secreted forms. Endoglycosidase H treatment of ruptured microsomes should remove about 2.3 kDa for each core structure cleaved. Fragment sizes for the Ctm HAh6αβla fusions are shown after removal of 29 kDa βlactamase by Kex2p cleavage.
In vitro expression of the PrPHAh6 constructs
T7 polymerase run-off transcripts of HAh6 constructs were translated in rabbit reticulocyte extracts in the presence of dog pancreas microsomes and the products analyzed, essentially as described by Hegde et al.Citation5 As shown for MH2M PrP, its A3V mutant and the KH-II mutant of SHa PrP (, lanes 1, 5, 8), expression of these fusions produced species close to the predicted 27 and 32 kDa sizes (), but due to a gel loading artifact, these appear as close pairs of bands. In all cases, the 32 kDa species was absent in the presence of NYT tripeptide, confirming its glycosylation status (lanes 2, 6, 9); mobility is consistent with glycosylation at both sites in PrP. The total glycosylated species (Ctm plus secreted) comprised about 20% of the total for MH2M and SHa (not shown) fusions and about 50% for the A3V and KH-II fusions. The previously reported effects of mutations increasing TM1 hydrophobicityCitation5 are, therefore reproduced and are not substantially affected by replacement of the GPI anchor with the HAh6 reporter.
Figure 3. In vitro expression of PrP-HAh6 fusions. T7 run-off transcripts were translated by rabbit reticulocyte extracts in the presence of dog pancreas microsomes in the presence or absence of NYT, a competitive inhibitor of glycosylation (lanes 1–2, 5–6, 8–9). Protease K treatment was performed in the presence (lane 4) or absence (lanes 3, 7, 10) of 1% triton × 100.
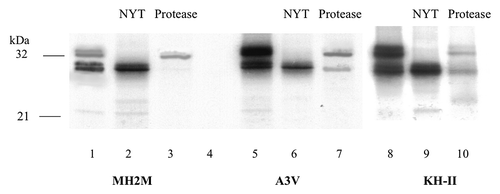
All species were rapidly destroyed by protease in the presence of Triton (lane 4), so no significant quantities of protease-resistant aggregates were present. In intact microsomes protease K hydrolysis should remove the HA tag from Ntm species; treatment eliminated the 27 kDa band from MH2M (lane 3) and SHa (not shown) fusion products, consistent with Ntm insertion. Protease K treatment should also distinguish secreted glycosylated products, which should remain intact, from Ctm TM forms, which should lose 7–8 kDa of N-terminal peptide (), producing a 24 kDa fragment. Protection against proteolysis by microsomal membranes is typically incomplete and this species was not clearly visible in MH2M products (lane 3). However, the predicted Ctm species fragment, expected to be more abundant in products from A3V and KH-II constructs, is visible in lanes 7 and 10. The protease-resistant 32 kDa band was of similar strength in lanes 3, 7 and 10, suggesting that secretion of PrP was not significantly affected by the A3V and KH-II mutations. Relative levels of Ctm and secreted species can only be estimated very roughly from band densities because of incomplete protease protection, but these and similar data indicate that Ctm accounts for 5–10% of the MH2M and SHa species and 30–40% of the KH-II and A3V species while secreted species are 10–20% in all.
Yeast expression of PrP-HAh6 fusions produces patterns similar to those seen in vitro
The HAh6 fusions of SHa, KH-II, MH2M and A3V-HAh6 PrP’s were expressed in a pep4-null mutant of yeast strain CRY2.Citation16 The pep4 mutation inactivates most vacuolar proteases, since Pep4p is required for their activation. This did not noticeably affect the in vivo stability of the PrP fusions but prevents the proteolysis that commonly accompanies cell breakage, even in the presence of a full complement of protease inhibitors (not shown). Spheroplasts were broken either by gentle rupture, intended to maintain microsomal integrity, or by vortexing with glass beads. Western blot data for 10 μg total protein from spheroplasts of about 3.10Citation6 cells expressing SHa and KH-II PrP’s are shown in . The 3000 g pellet (LSP) contains larger fragments and any residual intact cells and about 7% of the total HA fusion species detected (lanes 1). The 27 kDa band in this and other fractions corresponds in size to that predicted for the Ntm species lacking the 2.5 kDa signal peptide (). When PrP’s are expressed in yeast at much higher levels, most accumulate as aggregated, cytoplasmic species retaining the signal peptide.Citation17 This is apparently avoided at the expression levels described here. Most of the PrP fusion products are in the low-speed supernatant (LSS) containing membrane vesicles and cytoplasm (lanes 2). After subsequent fractionation at 250 000 g, most is found in the high-speed pellet (HSP) membrane fraction (lanes 4) rather than in the supernatant (HSS) cytoplasmic fraction (lanes 3). Treatment of lysed HSP fractions with endoglycosidase H converted the slower 32 kDa bands, which therefore represents N-glycosylated species, into a species migrating with the faster, unmodified (and therefore not glycosylated) species (lanes 5). This shift in mobility is consistent with a Ctm TM species, core N-glycosylated at both C-terminal sites (), as seen in vitro (). This also implies failure of the Ctm fusions to reach the yeast Golgi compartment, where long outer glycan chains are added. Bands of intermediate mobility, indicating incomplete N-glycosylation, were not visible in the SHa and KH-II products (), but can be seen in a better-resolved gel of the A3V products (see below). Nevertheless, glycosylation in yeast at both sites in PrP was apparently quite efficient.
Figure 4. (A) Yeast expression of SHa-HAh6 and its KH-II mutant. Spheroplasts were isolated from exponential phase cells and separated at 3 000 g into low-speed pellet (LSP) and supernatant (LSS) fractions. The LSS was then separated at 250 000 g into high-speed pellet (HSP, membranes) and supernatant (HSS, cytoplasm) fractions. The lysed HSP sample was hydrolyzed with endoglycosidase H (Endo H). Samples representing constant cell number were fractionated by SDS-PAGE and detected by western blot using anti-HA monoclonal antibody. (B) Membrane localization of MH2M- and A3V-HAh6 fusions. Samples were fractionated by SDS-PAGE and detected as in A. Spheroplasts were isolated from exponential phase cells and separated at 3000 g into low-speed supernatant (lanes 1, LSS) and pellet (lanes 2, LSP) fractions. Spheroplasts in the LSS were lysed and hydrolyzed with endoglycosidase H (Endo H, lanes 3). The LSS was separated at 250 000 g into supernatant (HSS, cytoplasm) and pellet (HSP, membranes) in buffer A1 (lanes 8–9), with the addition of 1% triton × 100, 0.1% sarkosyl (lanes 4–5) or in 0.1M pH 11.5 carbonate buffer (lanes 6–7). Lanes 10 show concentrated culture supernatant. Sample loads in lanes 6–10 were 5-fold higher than in lanes 1–5.
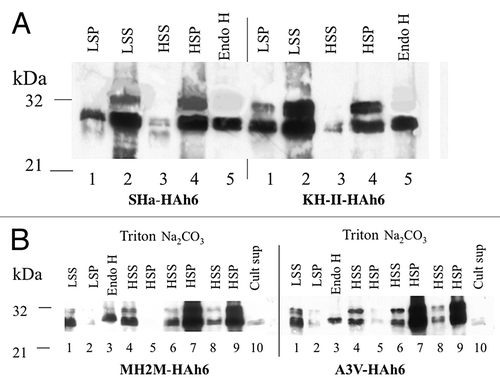
The MH2M- and A3V-HAh6 fusion products are shown in . Again, most are found in the LSS in intact spheroplasts (lanes 1) and in the HSP membrane fractions rather than the HSS cytoplasmic fractions (lanes 8 and 9). Almost none is found in a sample of concentrated culture supernatant representing secreted species from 5 fold higher cell numbers (lanes 10). Hydrolysis of lysed HSP fractions with endoglycosidase H again identifies the 32 kDa species as the glycosylated Ctm species (lanes 3). When lysed LSS membrane fractions are centrifuged at 250 000 g in the presence of 0.1M pH 11.5 carbonate buffer, the proteins remain in the HSP (lanes 6 and 7) while in the presence of 1% triton × 100, 0.1% sarkosyl (lanes 4 and 5), almost all are found in the HSS. Thus the 27 and 32 kDa Ntm and Ctm species both behave as non-aggregated, integral membrane proteins. This was also true for the SHa and KH-II fusion products (not shown). It is apparent that the KH-II and A3V mutations significantly increase Ctm TM insertion in yeast cells, as observed in vitro (; see below).
PrP-HAh6 fusions are primarily located in perinuclear regions
Spheroplasts expressing SHa-, A3V-, MH2M- and KH-II-HAh6 fusions were fixed with glutaraldehyde and stained with anti-HA and rhodamine-labeled anti-mouse antibodies. Most of the fusion proteins were located in annuli surrounding the DAPI-stained nuclei, with some in peripheral puncta (not shown). No significant staining was seen in the plasma membrane. This location, consistent with retention of these TM PrP species in the ER or associated membranes, appears to be similar, therefore to that reported for TM species in mammalian cells.Citation4
In vivo half-lives of both PrP fusion species are about 2 h
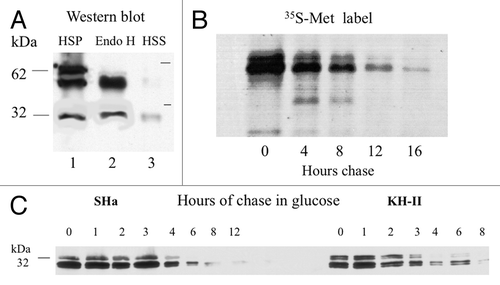
35S-Met pulse-chase analysis of half-lives was performed using the A3V-HAh9βla fusion, because the βla reporter doubles the Met content. To provide mobility controls, spheroplasts from an unlabeled culture were lysed, HSP and HSS fractions isolated and products analyzed by western blot using monoclonal anti-β-lactamase serum for detection. 63 and 58 kDa bands were detected ( lane 1), as predicted for Ctm and Ntm TM fusion proteins, respectively (). A band of about 29 kDa, presumably free β-lactamase, was also detected, presumably produced by turnover of the fusion. The HSS cytoplasmic fraction (lane 3) contained low levels of β-lactamase but very little of the fusion protein. Treatment of the HSP fraction with endoglycosidase H (Endo H) converted the slower band, which therefore represents N-glycosylated species, into a species migrating with the faster, unmodified (and therefore not glycosylated) species (lane 2). A similar pattern was seen for the KH-II-HAh9βla fusion (not shown). As for HAh6 fusions, the A3V-HAh9βla fusions appear to be membrane associated and the glycosylated, presumably Ctm fraction represents about 50% of the total and has the mobility predicted for core N-glycosylation at two sites, lacking Golgi N-glycosyl modification. Exponential growth phase cells of a parallel culture were labeled for 20 min with 35S-Met and chased with excess cold methionine. Products immunoprecipitated with anti-β-lactamase serum were detected by autoradiography after 4–16 h (). Densitometry of this and similar data sets for HAh6 fusions (not shown) indicated half-lives of about 2 h for both the slower, glycosylated and faster non-glycosylated species. The ratio of these bands, therefore, reflects their rates of synthesis.
Figure 5. (A) Yeast expression of A3V-HAh9βla. Cells were hydrolyzed with zymolyase and the low-speed supernatant spheroplast fraction separated at 250 000 g into pellet (HSP, lane 1) and supernatant (HSS, lane 3). Fractions were analyzed by SDS-PAGE and detected using monoclonal anti-β-lactamase serum. Lane 2 shows the products of endoglycosidase H treatment of the HSP fraction in lane 1. (B) pulse-chase half-life analysis. Cells in a parallel culture were labeled for 20 min with 35S methionine. After washing and chase with cold methionine for the indicated times, proteins immunoprecipitated with anti-β-lactamase serum were detected by autoradiography. (C) half-lives of SHa- and KH-II-HAh6 fusions determined by western blot. Exponential phase cells expressing SHa- or KH-II-HAh6 fusions from the GAL1 promoter were shifted from galactose to glucose medium at time zero. Samples were removed at the indicated times for western blot analysis using anti-HA monoclonal antibody. Band densities were determined by densitometry.
Western blot analysis confirms the 2 h PrP fusion half-lives
Western blot data can only be interpreted quantitatively if signals accurately reflect gel protein load. Response to load was tested by loading 1 to 12 μl of a membrane preparation from CRY2 cells expressing the A3V-HAh6 fusion. Signals following detection with anti-HA monoclonal antibody and a 2 or 10 min exposure time, as determined by densitometry, were precisely proportional to load over at least this 12-fold range, validating the use of western blots, with appropriate control of load and exposure, for determining the ratios of PrP fusion protein species. Replacement of the PGK promoter by the GAL1 promoterCitation18 in the MH2M, A3V, SHa and KH-II HAh6 fusions and growth of CRY2 transformants on galactose medium provided expression levels 2–3-fold higher than those seen on PGK in glucose media. Because a shift from galactose to glucose results in immediate and complete catabolite repression and rapid decay of GAL1 transcripts,Citation19 half-lives of PrP fusions can then be determined by decay of the signal in western blots. Half-lives for the 27 kDa, non-glycosylated and 32 kDa glycosylated species from expression of SHa and KH-II fusions () were both about 2 h, consistent with the pulse-label results for the HAh9βla fusions. Half-lives determined for the MH2M fusion species and its A3V mutant were similar (not shown). In conclusion, the ratios of PrP species detected in western blots should reflect their relative rates of synthesis.
Effects of variation in TM1 hydrophobicity on the ratio of Ntm and Ctm TM insertion in yeast
When HAh6 fusions were expressed in the in vitro mammalian system (), the fraction of secreted plus Ctm inserted species in the A3V and KH-II mutants was 2–3-fold higher than in the MH2M and SHa fusions (, row 1). Following yeast expression of HAh6 and HAh9βla fusions, the fraction of N-glycosylated Ctm plus secreted species, as determined by densitometry of western blots, (rows 2 and 3), were also approximately doubled for the A3V and KH-II fusions. Topology was also determined from the ratio of cell-associated and secreted β-lactamase activities in strain CRY2A cells expressing HAh6-αβla fusions.Citation15 Results were again similar (row 4). Average yeast cell data are shown in row 5. Thus, although higher levels of glycosylated species were produced in vitro (row 1), effects of the A3V and KH-II mutations were similar in vitro and in yeast cells.
Table 1. % N-glycosylated (Ctm + secreted) PrP fusion species
Expression of the MH2M- and A3V-HAh9βla fusions produces the predicted Ntm and Ctm TM species
Expression of the MH2M-HAh9βla fusion in strain CRY2 produced a single major 58 kDa band, detected in western blots using monoclonal anti-β-lactamase sera, all of which was located in the LSS and HSP microsomal fraction from gently lysed spheroplasts (, lane 1). Its mobility is that predicted for the Ntm TM form. This remained in the HSP following glass bead breakage in pH 11.5 carbonate buffer, but was in the HSS after breakage in 1% sarkosyl (not shown), so behaves as an integral membrane protein. It was converted by protease K treatment of intact microsomes in ice to a 29 kDa species corresponding in size to free β-lactamase (lane 2), consistent with Ntm insertion orientation. The A3V HAh9βla fusion (lane 4) produced both the 58 kDa Ntm species and a band of about 63 kDa, the size predicted for the core N-glycosylated Ctm species, as previously observed (). Proteolysis of intact microsomes (lane 5) produced both 29-kDa β-lactamase, presumably from Ntm species, and a 55 kDa band whose size is that predicted for the N-terminally truncated Ctm species (). This was trimmed to the predicted size of 50 kDa by endoglycosidase H (lane 6), consistent with derivation from the N-glycosylated Ctm TM species. No fusion protein was detectable in the HSS supernatant from 250 000 g fractionation of lysed microsomes (not shown), so none of the N-glycosylated species in this fusion appears to be secreted. As shown below, the K/E mutants acted as positive controls for PrP secretion in yeast. Analysis of the SHa- and KH-II-HAh9βla fusions gave results very similar to the MH2M and A3V fusions, respectively (not shown).
Figure 6. Effects of A3V, R136D, P102L, K106D, K110D and TSE25 mutations on translocation of MH2M fusions. (A) Cells expressing the indicated mutants of MH2M-HAh9βla were converted to spheroplasts, gently lysed, and microsomal membranes (3000 g supernatants, 250 000 g pellets) were isolated. Proteins were detected by western blot, using anti-β-lactamase serum after no treatment (lanes 1, 4, 7, 10, 13, 16), after exposure to protease K (lanes 2, 5, 8, 11, 14, 17), or after subsequent hydrolysis with endoglycosidase H (lanes 3, 6, 9, 12, 15, 18). β-lactamase released by protease K is detected as a 29 kDa species. (B) Cells expressing the indicated mutants of MH2M-HAh6 were broken with glass beads and proteins in the low speed supernatant were detected by western blot using anti-HA monoclonal antibody. Lanes 19–23 are all derived from the same blot. Lane 24 is from a separate blot run under identical conditions.
P102L and a KKR-TSE25 mutation at the mature N-terminus do not effect TM insertion
A P102L mutation in HuPrP causes Gerstmann-Sträussler-Scheinker disease and it seemed possible that enhanced α helicity in the charged region up stream of TM1 might enhance the effect of ΔCh. However, as shown in (lanes 10–12), this mutation did not detectably enhance Ctm insertion of the HAh9βla fusion in yeast. The KKRPK motif at the mature PrP N-terminus () is apparently important for controlling post-ER trafficking and lysozomal degradation of misfolded C-terminal PrP mutant proteins.Citation4 It is also principally responsible for a +2 ΔCh across the secretion signal peptide that might impair signal function by impeding the Ctm insertion required for translocation of downstream sequence, potentially resulting in signal retention and post-translational Ctm insertion of TM1. To test this, KKR25 in the MH2M-HAh6 fusion was mutated to TSE (), reversing signal peptide ΔCh to −2. Expression of the parent MH2M fusion produced predominantly the 27 kDa Ntm species (, lane 19), as previously shown (). Expression of the A3V-HAh6 fusion (lane 20) produced both the Ntm species and the 32 kDa Ctm species, again as previously shown (). A weak band at about 30 kDa, presumably corresponding to a minor fraction of mono-glycosylated Ctm species, was also visible on this gel. This band was not visible in the lower resolution gels shown in Figure 4B. The TSE mutant expressed predominantly the 27 kDa Ntm species (lane 24), like the MH2M fusion.
An R136D mutation in MH2M increases Ctm insertion to 35%
Deletion of the 103–110 segment, including 3 of the 4 positively charged residues immediately preceding TM1 and the first two TM1 residues, results in complete PrP secretion by microsomes in vitro.Citation5 This deletion significantly reduces TM1 length and hydrophobicity and reduces ΔCh from −5 to −2 and it is not clear whether either or both of these alterations is responsible. The R136D mutation in MH2M was designed to test the effect of increasing ΔCh from −5 to −7 without altering length or hydrophobicity. Expression of the HAh9βla fusion in strain CRY2 resulted in about 40% expression as the glycosylated Ctm species (, lane 7) and protease K treatment converted this to the 29 and 55 kDa species presumably derived, respectively, from Ntm and Ctm species, as for the A3V fusion (lane 8). The 55 kDa band was converted by EndoH to 50 kDa, as predicted for Ctm insertion (lane 9). Expression of the R136D-HAh6 fusion produced a similar increase in the 32 kDa Ctm species (, lane 21).
A K106E mutation in MH2M results in about 40% secretion and a K106E, K110E double mutant results in more than 60% secretion
A K106E mutation in the MH2M-HAh9βla background resulted in about 35% expression as the 63 kDa glycosylated species (, lane 13). In the K106/110E double mutant, glycosylation was increased to about 60% (lane 16). Similar levels of the 32 kDa N-glycosylated species are seen in cells expressing the K106E-HAh6 and double mutant fusions (, lanes 22, 23). A relatively faint but broad band of higher molecular weight species, presumably Golgi-modified secreted proteins, represented 5–10% of the proteins in lane 23 (not shown) so that the total of glycosylated species was at least 65%. Average data for the ratios of band densities from multiple blots (not including the high molecular weight band in the HAh6 fusions) are given in . The non-glycosylated 58 kDa species was destroyed by protease K treatment of intact microsomes, with appearance of the 29 kDa β-lactamase band, consistent with Ntm TM topology. However, much of the 63kDa species was unaffected (, lanes 14, 17), and this species was subsequently converted by EndoH to a species of about 58kDa, as predicted for secreted fusion protein (lanes 15, 18). Following protease K treatment, no 55 kDa band corresponding to Ctm TM insertion was detected (lanes 14, 17). It seems, therefore, that reducing ΔCh at TM1 from –5 to –3 partially relieves inhibition of PrP secretion in yeast, and secretion is about 65% when ΔCh is decreased to −1. Most of the secreted species, however, fail to receive Golgi modification, presumably remaining ER-associated.
Table 2. % N-glycosylated (Ctm TM + secreted) PrP fusion proteins expressed by mutants that alter ΔCh at TM1
Discussion
In vitro secretion efficiency for MH2M and SHa PrP’s by mammalian microsomes is reported to be about 50%, the rest being inserted as Ntm (40%) and N-glycosylated Ctm (10%) TM species.Citation5 Using HAh6 fusions lacking the GPI anchor, we observed distinctly lower (~15%) in vitro secretion efficiency, with a corresponding increase in Ntm insertion. However, the A3V and KH-II mutations, both increasing TM1 hydrophobicity, increased Ctm species to 35%, close to the 40% previously reported in vitro,Citation5 but with a corresponding decrease in Ntm species rather than fully secreted species as previously reported.Citation5 Thus replacement of the GPI anchor does not significantly alter in vitro topological responses to TM1 hydrophobicity.
In yeast cells, western blot analysis of the products of expression of the same MH2M and SHa PrP fusions on the same vector used for in vitro expression demonstrates the same Ntm and Ctm TM species. SDS-PAGE band mobilities and the effects on mobility of exposure of intact microsomes to protease K and of ruptured microsomes to endoglycosidase H are consistent with predictions for orientation, N-glycosylation and size of these TM species (, ). In mammalian cells, N-glycosylation of PrP lacking its GPI anchor is compromised to an extent dependent on the cell line employed. In an epithelial cell line, for example, about 70% is fully secreted but only 40% of this is fully glycosylated and most of the residual 30% is not glycosylated and remains in the Golgi.Citation20 By contrast, glycosylation in yeast cells appears to be quite efficient. At the moderate expression levels achieved with the pPGKT7g vector, translocation of the PrP fusions described is also efficient, avoiding the accumulation of cytoplasmic aggregates reported when PrP’s are expressed in yeast at much higher levels.Citation17 However, secretion is barely detectable for the parent MH2M and SHa fusions and the TM species produced appear to remain associated with the ER. The Ctm species, at least, does not reach the Golgi. Western blot analysis shows that both TM species have half-lives of about 2 h, so signals for accumulated PrP species reflect relative synthesis rates, with much greater ease and sensitivity than pulse-chase analysis, which gives similar ~2hr half-lives. Significantly, in spite of the failure of yeast cells to secrete the normal PrP peptide fusions, the A3V and KH-II mutations both increase the Ctm fraction to 35%, so effects on TM insertion orientation reflect those seen in vitro and in mouse brain. CtmPrP produced in mouse brain is fully glycosylated and GPI anchored, with a mature N-terminusCitation14 and results, therefore, from slippage of the N-terminus back to the cytoplasm following signal peptidase action and Ctm insertion of the TM1 segment. The size of Ctm products made in yeast are consistent with a mature N-terminus following loss of the 2.5 kDa signal peptide, but N-terminal sequence has not been determined.
In contrast to the results of in vitro and yeast expression, TM PrP species are barely detectable in transgenic mouse brain unless very sensitive techniques are employed.Citation14 This probably reflects more efficient translocation and the efficacy of normal quality control and turnover mechanisms. However, when the A3V and KH-II mutants are expressed in mice, Ctm forms become readily detectable,Citation5 presumably as these mechanisms become overburdened. Ctm PrP is also detected in human and mouse brains expressing the pathogenic A117V Prnp mutation, essentially a milder form of the A3V mutation.Citation5 Accumulating evidence for a correlation between neuropathogenesis and Ctm PrP production identifies this species, potentially in concert with cytoplasmic PrP (see below), as a strong candidate for a primary toxic agent.Citation3
TM1 hydrophobicity is marginal for membrane insertion.Citation21,Citation22 Its flanking sequences, KPSKPKTNMKH111 and SR136MMHFGNDWEDR (), have a net ΔCh of about −5, and electrostatic interaction with a net negative charge at the translocon entranceCitation23,Citation24 would favor Ctm translocon insertion,Citation24,Citation25 incompatible with PrP secretion. Any delay in translocation caused by this electrostatic interaction would promote lateral TM1 insertion, but insertion would be Ntm unless retro-translocation of the N-terminus allowed Ctm insertion. Increased TM1 hydrophobicity, not surprisingly, enhances TM insertion, but in the Ctm orientation directed by the local ΔCh. Deletion of residues 104–114, which both reduces ΔCh to −2 and also reduces TM1 length and hydrophobicity, results in complete PrP in vitro secretion.Citation5 By exchanging positively and negatively charged residues in the segments flanking TM1, effects of altering ΔCh without affecting length or local hydrophobicity were tested. The R136D mutation enhances ΔCh to −7 and resulted in an increase in Ctm insertion equivalent to that caused by the A3V and KH-II mutations. The K106E mutation decreases ΔCh to −3 and allowed 35% secretion of PrP in yeast while the double mutant with K110E, decreasing ΔCh to −1, allowed about 65% secretion. Yeast cells can export secreted and TM proteins by SRP-independent post-translational translocation.Citation26 However, if this were responsible for TM insertion of PrP, ΔCh would ensure predominantly Ctm insertion and decreasing ΔCh would only increase the Ntm/Ctm ratio, contrary to our results. We have, therefore, demonstrated that variation in TM1 hydrophobicity has the same effect on the orientation of TM PrP insertion in yeast cells as it does in mammalian cells and have demonstrated the utility of the yeast model by using it to demonstrate a prominent role for TM1 ΔCh in driving TM PrP insertion.
The inverse square law governing electrostatic interactions suggests that effects of ΔCh may be dependent on secondary structure in the TM flanking segments. It seemed possible that the P102L PrP mutation that, like A117V, causes Gerstmann-Sträussler-Scheinker syndrome, could be pathogenic because of influence on the effect of TM1 ΔCh. In yeast cells this would manifest as an increase in Ctm insertion. This mutation in the MH2M-HAh6 fusion, however, caused no significant change in Ctm/Ntm TM insertion ratio. The TSE mutation that reversed ΔCh across the secretion signal, potentially enhancing its function, had little apparent effect. Interaction between PrP signal function and TM1 insertion is complex.Citation9
A minimal reconstituted proteoliposome system, competent for translocating many secreted proteins, has very low efficiency for PrP translocation unless the PrP secretion signal is replaced by a more efficient sequence that promotes formation of a tight ribosome-translocon seal.Citation10 The ribosome-associated TRAP complex was identified as a cofactor enhancing secretion of PrP and other proteins with suboptimal signal sequences.Citation27 Yeast has no counterpart to TRAP, presumably explaining its inability to secrete the normal PrP sequence. Since reducing ΔCh does allow secretion in yeast, TRAP may have a related function in mammalian cells, suppressing the translocation delay produced by a negative ΔCh associated with a potential TM segment, perhaps by masking negative charge at the entrance to the Sec61 channel. This might also mitigate effects of a positive ΔCh on signal function. Since response to such ΔCh signals is faithfully executed in types II and III TM proteins, TRAP function may be selectively recruited to N-terminal signal/SRP receptor/Sec61 nascent secretion complexes.
It is hypothesized that an incomplete ribosome-translocon seal, produced by interaction of the PrP secretion signal with the translocon, allows translocation to cease when the ER folding machinery is stressed, resulting in cytoplasmic accumulation of nascent peptide.Citation10,Citation28 PrP is a relatively abundant protein species in neurons, and it is suggested that cytoplasmic accumulation may facilitate proteasomal degradation and so be less acutely toxic than accumulation in the ER lumen,Citation29 perhaps providing selection for a sub-optimal secretion signal. The conserved charges flanking TM1 may contribute to such a mechanism. Cytoplasmic accumulation of PrP occurs in mouse models of pathogenic C-terminal PrP mutants that do not cause CtmPrP to increase.Citation30 Other mutant misfolded PrP’s appear to be poor substrates for retro-translocation from the ER and accumulate in post-ER Golgi-like vesicles.Citation4 In mice, loss of Mahogunin Ring Finger 1 E3 ubiquitin ligase causes spongiform neurodegeneration,Citation31 probably by disrupting endosome-lysosome trafficking, and both CtmPrP and cytosolic PrP sequester Mahogunin, disrupting its function, potentially a primary cause of neurotoxicity.Citation32 Alternatively, Cu2+ bound to the octad repeat motifs in the PrP N-terminus,Citation7 in the presence of NO released by inflammatory responses in adjacent activated microglia, is potentially capable of catalyzing nitrosylation of free cysteine sulfhydryl groups in nearby proteins.Citation33 Since these are predominantly present in the reducing environment of the cytoplasm, Ctm and cytoplasmic PrP species are most capable of generating such modified proteins. ER-associated Ctm species, in particular, may be best located to modify the major transient population of free sulfhydryl groups, those in nascent proteins traversing the translocon, destined to form cystine cross links in the ER lumen. Such nitrosylation events, by chronically enhancing the unfolded ER protein and proteasomal burdens, stimulating translation arrest through the Unfolded Protein Response, could be an alternative driver of neurotoxicity. Thus, while the role, if any, of NtmPrP in pathogenicity is unknown, the chronic accumulation of either CtmPrP or cytoplasmic PrPCitation34 may be a proximal neurotoxic event in TSE’s induced by PrPSc production.
Materials and Methods
Yeast strains used for expression of PrP fusions
Strain CRY2 ura3–1, leu2–3, 112, his3–11 trp-1Citation35 was modified by replacement of the PEP4 gene by HIS3, resulting in inactivation of Pep4p, vacuolar proteinase A. Most vacuolar proteases are inactivated since these require Pep4p for activation; proteolytic degradation of proteins during cell rupture is greatly reduced. In strain CRY2A, ER-Kex2p, under control of the PGK promoter was inserted in strain CRY2 Δpep4 at the his3–11 locus.Citation16 Expression of Kex2p activity in the ER is then constitutive; this strain was used for analysis of secreted β-lactamase from expression of HAh6-αβla fusions.
Vectors for expression of PrP’s both in vitro and in yeast cells
pPGKT7g
pS79αB and pS79PBCitation12 are YEp URA3 vectors developed for the analysis of TM protein topology in yeast.Citation12,Citation15,Citation16,Citation36 S79, the 79 residue N-terminal fragment of the Ste2p yeast pheromone receptor, a model TM protein, is fused to either of two C-terminal reporters. In the αB reporter, α, a 34 residue fragment of pre-pro-αfactor, including two N-glycosylation sites and a terminal KR site for cleavage by Kex2p, is followed by B, the mature sequence of β-lactamase. In PB, α is replaced by P, a larger fragment of yeast type 1 pre-pro-toxin providing similar sites. These fusions are located within an expression cassette consisting of the pPGK promoter and tPGK terminator, providing constitutive high level expression in yeast.Citation12 In pGAL-S79αB, the pGAL1 promoter fragment replaces the Hind3 to Xho1 pPGK fragment in pS79αB. A 46 bp sequence encoding the T7 promoter (underlined), followed by the 18bp pre-sequence of rabbit globin, was inserted by annealing the kinased oligomers (5′) TCGAGCTCTA ATACGACTCA CTATACGACT TGCAATCCCC CAAAAC and (5′) TCGAGTTTTG GGGGATTGCA AGTCCTATAG TGAGCTGTAT TAGAGC and ligation to pS79PB cut at the unique Xho1 site upstream of the translation start site and treated with phosphatase. The product is pPGKT7g expressing S79PB. Insertion orientation was confirmed by sequence analysis.
pPGKT7g-HAh6
The triple HA epitope tag was cloned by PCR as a 104 bp Xho1/Nsi1 to Nhe1/Bgl2 fragment using primers (5′) GGGCTCGAGA TACATGCATC GTACCCATAC GATGTTCC and (5′) GGGAGATCTT AGCTAGCGTA ATCTGGAAC (restriction sites are in bold). Cutting with Xho1 and Bgl2 and insertion in pPGKT7g cut with the same enzymes deleted S79PB and produced pPGKT7g-HA. Oligomers (5′) CTAGCCATCA TCATCATCAT CATTAA and GATCTTAATG ATGATGATGA TGATGATG were kinased, annealed and ligated to pPGKT7g-HA cut Nhe + Bgl2 producing pPGKT7g-HAh6.
PrP-HAh6 fusions
Plasmids expressing the Syrian Hamster (SHa) and MH2M PrP’s and their KH-II and A3V mutants from the SP6 promoter were provided by Dr. Lingappa.Citation5 MH2M is a chimera in which residues 95 to 188 of SHa PrP have been inserted into the equivalent location in Mouse PrP. As a result, residues 57 to 204, including TM1 and its flanking sequences, are identical in SHa and MH2M. Primers (5′) GGGCTCGAGAAAATGGCGAACCTTGGCTACTGGGT and (5′) GGGCTGCAGA ATTCTGCATG CTCATTCTAG AGCTCCTTCT CCCGTCGTAA TAGGCCTG were used to copy MH2M and A3V codons 1–232 from these clones. The products were inserted into pPGKT7g-HAh6, cut Xho1 + Nsi1 as Xho1 to Pst1 fragments. The products are MH2M- and A3VHAh6 in which the GPI addition signals are replaced by the HAh6 tag. Primers (5′) GGGCTCGAGA AAATGGCGAA CCTTAGCTAC TGGCT and (5′) GGGCTGCAGA ATTCTGCATG CTCATTCTAG AGCTCCTTCT TCCATCGTAG TAGGCCT were used to copy SHa and KH-II codons 1–232 from the clones provided by Dr Lingappa.Citation5 The products were inserted into pPGKT7g-HAh6, cut Xho1 + Nsi1 as Xho1 to Pst1 fragments producing SHa- and KHII-HAh6.
PrP-HAh6-αβla fusions
β-lactamase assays for topology determination
Primers (5′) GGGGCTAGCC ATCATCATCA TCATCATGTT TCCAACAGCA CAAATAACG and (5′) GGGAGATCTT ACCAATGCTT AATCAGTGAG were used to clone αβla by PCR from pGAL-S79αB.Citation12 The product was cut with Nhe1 and Bgl2 for insertion into the above HAh6 constructs to produce HAh6-αβla fusions. For topology analysis these constructs were transformed into strain CRY2A. In this strain, The Kex2p protease, normally located in the lumen of the Golgi, is also present in the ER lumen. Efficient cleavage of both secreted and Ctm TM fusions releases βlactamase for secretion (, bottom).Citation16 Ntm fusions remain intact, so the ratio of cell-associated to secreted activity reflects the ratio of Ntm to (Ctm + secreted) species.Citation12,Citation15,Citation16 β-lactamase assays were performed as previously described.Citation18
PrP-h9βla fusions
Primers (5′) GGGGCTAGCC ATCATCATCA TCATCATCAT CATCATACTA GTCACCCAGA AACGCTGG and (5′) GGGAGATCTT AACCAATGCT TAATCAGTGA G were used to clone β-lactamase preceded by 9 His codons using pGAL-S79αB as template. The product was cut with Nhe1 and Bgl2 and cloned into Prp-HAh6 fusions cut with Nhe1 and Bgl2 producing PrP-h9βla fusions lacking any site for cleavage by Kex2p so that Ctm and secreted products should remain intact in the ER lumen.
Mutagenesis of MH2M fusions
57bp oligomers were used to construct the K106E K110E single and double mutants. These were kinased, annealed and used to replace the Acc651 to Ngo4 fragment of MH2M-HAh6. An Nde1 site is introduced. Forward: (5′) GTACCCACAA TCAGTGGAAC AAGCCCAGTA AGCCAGAAAC CAACATGRAG CATATGG. Reverse (5′) CCGGCCATAT GCTYCATGTT GGTTTCTGGC TTACTGGGCT TGTTCCACTG ATTGTGG. R is A or G and Y is T or C. Sequence of independent clones identified the Glu (GAA) codon at 106 and distinguished Lys (AAG) from Glu (GAG) at residue 110. A similar pair of oligomers produced a 57 bp Acc651 to Ngo4 fragment to introduce the P102L mutation and a Hind3 site (AAG-CCC to AAG CTT). The following PCR primer pairs were used to mutate pPGKT7g-MH2M-HAh6. The R136D mutation and a BamH1 site were introduced using mutagenic primers (5′) GGGAGTGCCA TGTCGGATCC CATGATGCAT TTTGG and (5′) CCAAAATGCA TCATGGGATC CGACATGGCA CTCCC. Mutagenic primers (5′) GATGTCGGCC TCTGCACTAG TGAGCCAAAG CCTGGAG and (5′) CTCCAGGCTT TGGCTCACTA GTGCAGAGGC CGACATC were used to mutate the PrP mature N-terminus from Lys Lys Arg to Thr Ala Glu. A Spe1 site is introduced. Products were cloned in XL-1 blue cells and identified by the altered restriction sites (bold). Sequences were confirmed.
In vitro translation of T7 transcripts
pPGKT7g plasmid constructs expressing MH2M and SHa PrP HAh6 fusions and their A3V and KH-II mutants were linearized by cleavage at the unique EcoR1 site downstream of the tPGK terminator. T7 polymerase run-off transcripts were translated in the presence of 35S-Methionine using rabbit reticulocytes in the presence of dog-pancreas microsomes, both kindly provided by Dr Reid Gilmore. The products were immunoprecipitated using monoclonal F7 anti-HA antibodies (Santa Cruz biotechnology Inc.), fractionated by SDS-PAGE, detected by autoradiography and relative band intensities determined by densitometry.
Culture and analysis of Yeast transformants
pPGKT7g vector transformants of strain CRY2 or CRY2A were selected on Ura D/O medium and independent clones were tested for expression as described below. Stock cultures were diluted 1000 fold in 2% glucose Ura D/O medium and grown overnight at 30 °C. pGAL vector transformants for half life analysis were initially grown in 2% galactose Ura D/O medium before transfer to glucose medium to terminate expression and initiate chase. Cells for analysis of protein content by disruption were harvested in late exponential phase and washed in buffer A1 (13) containing EDTA, PMSF and pepstatin protease inhibitors. About 108 cells were suspended in ice in 0.2 ml buffer A1 with 0.2 ml 0.5 mm beads and vortexed 8 × 40 s with cooling in ice between breaks. Recovered material was centrifuged for 3 min at 3000 g producing LSS and LSP fractions (fractionation by this first simple step is no more than 95% complete). The LSP was suspended in 0.2 ml buffer A1 for analysis and the LSS was spun for 20 min at 4 °C in an airfuge at 250 000 g to separate membrane pellet (high speed pellet HSP) and supernatant (HSS) fractions.
For preparation of intact microsomes
108 cells were suspended in 0.2 ml buffer A (without protease inhibitors) + 1.4 M sorbitol + 10 ug zymolyase 100T (Sigma-Aldrich) and 2 μl β-mercaptoethanol. After about 45 min at 23 °C, when lysis of a sample in 1% sarkosyl was observed to be complete, debris was removed by centrifugation at 4000 g for 10 min (low speed pellet, LSP). The supernatant (LSS) was spun in an airfuge at 250 000 g for 10 min, separating the cytoplasmic HSS and HSP microsome pellet fractions. The HSP was gently suspended in 0.2 ml buffer A + 1.4 M sorbitol for analysis.
For protease K hydrolysis of intact microsomes
100 μl of the microsome suspension was mixed with 0.1 μg proteinase K and 5 mm CaCl2. After 90 min on ice, 3 mM PMSF was added to stop the reaction.
For endoglycosidase H hydrolysis
Microsomes were pelleted at 250 000 g, washed by suspension in buffer A1 (with protease inhibitors) + 1.4 M sorbitol and pelleting again, and lysed by suspension in 0.1 M NaOAc buffer pH 5.5 + 1% triton × 100 and 0.1% sarkosyl. Samples (25 μl) were mixed with endoglycosidase H (10 μl, Boehringer 11088726) and incubated for 2 h at 37 °C.
SDS-PAGE and western blot analysis
Protein samples in sample buffer (10% glycerol, 2% SDS, 62 mM Tris pH 6.8, 2% β-mercaptoethanol) were heated 3 min at 100 °C and loaded onto 10 or 12.5% gels in a Miniprotean apparatus (BioRad), run at 150 V for 1.25 h, blotted onto immobilon membranes, blocked with 5% non-fat dry milk and detected with monoclonal F7 anti-HA (Santa Cruz biotechnology Inc.) or mouse monoclonal anti-β-lactamase 8A5-A10 IgG1 (Biodesign International) as the primary antibody followed by HRP (peroxidase)-goat anti-mouse secondary antibody and detection using a SuperSignal West pico kit (Thermo Scientific).
35S-Met pulse labeling and chase
108 mid log-phase cells of strain CRY2 transformants in Ura/Met D/O medium (1 ml) were labeled with 0.2 mCi of Perkin Elmer NEG-072 35S-protein labeling mix for 20 min. After addition of 0.1 ml 20 mM L-Met, cells were pelleted and regrown in 10 ml UraD/O chase medium. Samples (2 ml) were removed at intervals, extracted proteins immunoprecipitated, fractionated by SDS-PAGE and detected by autoradiography.
| Abbreviations: | ||
| PrP | = | prion protein |
| GPI | = | glycophosphatidyl inositol |
| TM | = | transmembrane |
| ER | = | endoplasmic reticulum |
| Ctm | = | C-terminus translocated to ER lumen |
| Ntm | = | N-terminus translocated to ER lumen |
| ΔCh | = | charge difference across a TM segment |
| SRP | = | signal recognition particle |
| TSE | = | transmissible spongiform encephalopathy |
| TRAP | = | translocon-associated protein |
| HA | = | hemagglutinin epitope |
| βla | = | β-lactamase |
| LSS and LSP | = | low speed supernatant and pellet |
| HSS and HSP | = | high speed supernatant and pellet |
| EndoH | = | endoglycosidase H |
| Met | = | methionine |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by grant A2001-080 from the American Health Assistance Foundation and F54 from the University of Massachusetts President’s S&T Initiatives fund. We would like to thank Dr Lingappa for gifts of plasmids and Dr Reid Gilmore for gifts of T7 RNA polymerase and dog pancreas membrane vesicles, for helpful advice and for critical reading of the manuscript.
References
- Prusiner SB. Prions. Proc Natl Acad Sci U S A 1998; 95:13363 - 83; http://dx.doi.org/10.1073/pnas.95.23.13363; PMID: 9811807
- Brandner S, Isenmann S, Raeber A, Fischer M, Sailer A, Kobayashi Y, Marino S, Weissmann C, Aguzzi A. Normal host prion protein necessary for scrapie-induced neurotoxicity. Nature 1996; 379:339 - 43; http://dx.doi.org/10.1038/379339a0; PMID: 8552188
- Chakrabarti O, Ashok A, Hegde RS. Prion protein biosynthesis and its emerging role in neurodegeneration. Trends Biochem Sci 2009; 34:287 - 95; http://dx.doi.org/10.1016/j.tibs.2009.03.001; PMID: 19447626
- Ashok A, Hegde RS. Selective processing and metabolism of disease-causing mutant prion proteins. PLoS Pathog 2009; 5:e1000479; http://dx.doi.org/10.1371/journal.ppat.1000479; PMID: 19543376
- Hegde RS, Mastrianni JA, Scott MR, DeFea KA, Tremblay P, Torchia M, DeArmond SJ, Prusiner SB, Lingappa VR. A transmembrane form of the prion protein in neurodegenerative disease. Science 1998; 279:827 - 34; http://dx.doi.org/10.1126/science.279.5352.827; PMID: 9452375
- Liu H, Farr-Jones S, Ulyanov NB, Llinas M, Marqusee S, Groth D, Cohen FE, Prusiner SB, James TL. Solution structure of Syrian hamster prion protein rPrP(90-231). Biochemistry 1999; 38:5362 - 77; http://dx.doi.org/10.1021/bi982878x; PMID: 10220323
- Brown DR. Copper and prion diseases. Biochem Soc Trans 2002; 30:742 - 5; http://dx.doi.org/10.1042/BST0300742; PMID: 12196183
- Pauly PC, Harris DA. Copper stimulates endocytosis of the prion protein. J Biol Chem 1998; 273:33107 - 10; http://dx.doi.org/10.1074/jbc.273.50.33107; PMID: 9837873
- Kim SJ, Rahbar R, Hegde RS. Combinatorial control of prion protein biogenesis by the signal sequence and transmembrane domain. J Biol Chem 2001; 276:26132 - 40; http://dx.doi.org/10.1074/jbc.M101638200; PMID: 11359769
- Rane NS, Chakrabarti O, Feigenbaum L, Hegde RS. Signal sequence insufficiency contributes to neurodegeneration caused by transmembrane prion protein. J Cell Biol 2010; 188:515 - 26; http://dx.doi.org/10.1083/jcb.200911115; PMID: 20156965
- Beltzer JP, Fiedler K, Fuhrer C, Geffen I, Handschin C, Wessels HP, Spiess M. Charged residues are major determinants of the transmembrane orientation of a signal-anchor sequence. J Biol Chem 1991; 266:973 - 8; PMID: 1985975
- Harley CA, Tipper DJ. The role of charged residues in determining transmembrane protein insertion orientation in yeast. J Biol Chem 1996; 271:24625 - 33; http://dx.doi.org/10.1074/jbc.271.40.24625; PMID: 8798728
- Hartmann E, Rapoport TA, Lodish HF. Predicting the orientation of eukaryotic membrane-spanning proteins. Proc Natl Acad Sci U S A 1989; 86:5786 - 90; http://dx.doi.org/10.1073/pnas.86.15.5786; PMID: 2762295
- Emerman AB, Zhang ZR, Chakrabarti O, Hegde RS. Compartment-restricted biotinylation reveals novel features of prion protein metabolism in vivo. Mol Biol Cell 2010; 21:4325 - 37; http://dx.doi.org/10.1091/mbc.E10-09-0742; PMID: 20980618
- Cartwright CP, Tipper DJ. In vivo topological analysis of Ste2, a yeast plasma membrane protein, by using beta-lactamase gene fusions. Mol Cell Biol 1991; 11:2620 - 8; PMID: 2017168
- Harley CA, Holt JA, Turner R, Tipper DJ. Transmembrane protein insertion orientation in yeast depends on the charge difference across transmembrane segments, their total hydrophobicity, and its distribution. J Biol Chem 1998; 273:24963 - 71; http://dx.doi.org/10.1074/jbc.273.38.24963; PMID: 9733804
- Ma J, Lindquist S. De novo generation of a PrPSc-like conformation in living cells. Nat Cell Biol 1999; 1:358 - 61; http://dx.doi.org/10.1038/14053; PMID: 10559963
- Cartwright CP, Li Y, Zhu YS, Kang YS, Tipper DJ. Use of beta-lactamase as a secreted reporter of promoter function in yeast. Yeast 1994; 10:497 - 508; http://dx.doi.org/10.1002/yea.320100409; PMID: 7941736
- Munchel SE, Shultzaberger RK, Takizawa N, Weis K. Dynamic profiling of mRNA turnover reveals gene-specific and system-wide regulation of mRNA decay. Mol Biol Cell 2011; 22:2787 - 95; http://dx.doi.org/10.1091/mbc.E11-01-0028; PMID: 21680716
- Biswas S, Langeveld JP, Tipper D, Lu S. Intracellular accumulation of a 46 kDa species of mouse prion protein as a result of loss of glycosylation in cultured mammalian cells. Biochem Biophys Res Commun 2006; 349:153 - 61; http://dx.doi.org/10.1016/j.bbrc.2006.08.035; PMID: 16935263
- Hessa T, Meindl-Beinker NM, Bernsel A, Kim H, Sato Y, Lerch-Bader M, Nilsson I, White SH, von Heijne G. Molecular code for transmembrane-helix recognition by the Sec61 translocon. Nature 2007; 450:1026 - 30; http://dx.doi.org/10.1038/nature06387; PMID: 18075582
- Lundin C, Kim H, Nilsson I, White SH, von Heijne G. Molecular code for protein insertion in the endoplasmic reticulum membrane is similar for N(in)-C(out) and N(out)-C(in) transmembrane helices. Proc Natl Acad Sci U S A 2008; 105:15702 - 7; http://dx.doi.org/10.1073/pnas.0804842105; PMID: 18840693
- Junne T, Schwede T, Goder V, Spiess M. Mutations in the Sec61p channel affecting signal sequence recognition and membrane protein topology. J Biol Chem 2007; 282:33201 - 9; http://dx.doi.org/10.1074/jbc.M707219200; PMID: 17893139
- Bogdanov M, Xie J, Dowhan W. Lipid-protein interactions drive membrane protein topogenesis in accordance with the positive inside rule. J Biol Chem 2009; 284:9637 - 41; http://dx.doi.org/10.1074/jbc.R800081200; PMID: 19074771
- von Heijne G. Membrane protein structure prediction. Hydrophobicity analysis and the positive-inside rule. J Mol Biol 1992; 225:487 - 94; http://dx.doi.org/10.1016/0022-2836(92)90934-C; PMID: 1593632
- Reithinger JH, Kim JE, Kim H. Sec62 protein mediates membrane insertion and orientation of moderately hydrophobic signal anchor proteins in the endoplasmic reticulum (ER). J Biol Chem 2013; 288:18058 - 67; http://dx.doi.org/10.1074/jbc.M113.473009; PMID: 23632075
- Fons RD, Bogert BA, Hegde RS. Substrate-specific function of the translocon-associated protein complex during translocation across the ER membrane. J Cell Biol 2003; 160:529 - 39; http://dx.doi.org/10.1083/jcb.200210095; PMID: 12578908
- Hegde RS, Kang SW. The concept of translocational regulation. J Cell Biol 2008; 182:225 - 32; http://dx.doi.org/10.1083/jcb.200804157; PMID: 18644895
- Rane NS, Kang SW, Chakrabarti O, Feigenbaum L, Hegde RS. Reduced translocation of nascent prion protein during ER stress contributes to neurodegeneration. Dev Cell 2008; 15:359 - 70; http://dx.doi.org/10.1016/j.devcel.2008.06.015; PMID: 18804434
- Ma J, Wollmann R, Lindquist S. Neurotoxicity and neurodegeneration when PrP accumulates in the cytosol. Science 2002; 298:1781 - 5; http://dx.doi.org/10.1126/science.1073725; PMID: 12386337
- Whatley BR, Li L, Chin LS. The ubiquitin-proteasome system in spongiform degenerative disorders. Biochim Biophys Acta 2008; 1782:700 - 12; http://dx.doi.org/10.1016/j.bbadis.2008.08.006; PMID: 18790052
- Chakrabarti O, Hegde RS. Functional depletion of mahogunin by cytosolically exposed prion protein contributes to neurodegeneration. Cell 2009; 137:1136 - 47; http://dx.doi.org/10.1016/j.cell.2009.03.042; PMID: 19524515
- Nakamura T, Tu S, Akhtar MW, Sunico CR, Okamoto S, Lipton SA. Aberrant protein s-nitrosylation in neurodegenerative diseases. Neuron 2013; 78:596 - 614; http://dx.doi.org/10.1016/j.neuron.2013.05.005; PMID: 23719160
- Orsi A, Fioriti L, Chiesa R, Sitia R. Conditions of endoplasmic reticulum stress favor the accumulation of cytosolic prion protein. J Biol Chem 2006; 281:30431 - 8; http://dx.doi.org/10.1074/jbc.M605320200; PMID: 16908519
- Komano H, Fuller RS. Shared functions in vivo of a glycosyl-phosphatidylinositol-linked aspartyl protease, Mkc7, and the proprotein processing protease Kex2 in yeast. Proc Natl Acad Sci U S A 1995; 92:10752 - 6; http://dx.doi.org/10.1073/pnas.92.23.10752; PMID: 7479877
- Cartwright CP, Zhu YS, Tipper DJ. Efficient secretion in yeast based on fragments from K1 killer preprotoxin. Yeast 1992; 8:261 - 72; http://dx.doi.org/10.1002/yea.320080404; PMID: 1514325
- Hegde RS, Voigt S, Lingappa VR. Regulation of protein topology by trans-acting factors at the endoplasmic reticulum. Mol Cell 1998; 2:85 - 91; http://dx.doi.org/10.1016/S1097-2765(00)80116-1; PMID: 9702194