Abstract
Alzheimer disease is associated with the accumulation of oligomeric amyloid β peptide (Aβ), accompanied by synaptic dysfunction and neuronal death. Polymeric form of prion protein (PrP), PrPSc, is implicated in transmissible spongiform encephalopathies (TSEs). Recently, it was shown that the monomeric cellular form of PrP (PrPC), located on the neuron surface, binds Aβ oligomers (and possibly other β-rich conformers) via the PrP23–27 and PrP90–110 segments, acting as Aβ receptor. On the other hand, PrPSc polymers efficiently bind to Aβ monomers and accelerate their oligomerization. To identify specific PrP sequences that are essential for the interaction between PrP polymers and Aβ peptide, we have co-expressed Aβ and PrP (or its shortened derivatives), fused to different fluorophores, in the yeast cell. Our data show that the 90–110 and 28–89 regions of PrP control the binding of proteinase-resistant PrP polymers to the Aβ peptide, whereas the 23–27 segment of PrP is dispensable for this interaction. This indicates that the set of PrP fragments involved in the interaction with Aβ depends on PrP conformational state.
Introduction
Experimental evidence suggests that amyloid β (Aβ) oligomers play a crucial role in the pathogenesis of Alzheimer disease (AD).Citation1,Citation2 Accumulation of Aβ oligomers leads to neurotoxicity and impairs synaptic plasticity and long-term memory.Citation3,Citation4 However, the mechanisms by which Aβ oligomers cause neuropathology remain unclear. One potential lead comes from the ability of Aβ oligomers to interact with certain receptors on the synaptic membrane,Citation5-Citation7 in particular, with the cellular isoform of prion protein (PrPC).Citation8,Citation9 Normal cellular function of PrPC is still unclear although its polymeric (prion) isoform, PrPSc, is a causative agent of transmissible prion diseases, the neurodenerative diseases with ethiology not dissimilar from AD.Citation10 PrPC is located on the neuron surface and is shown to interact with β-sheet-rich conformers of various origins, such as heterologous PrP, yeast prion proteins or engineered β-peptides, in addition to Aβ peptide.Citation11 Thus, PrPC acts as a universal receptor for amyloid-like oligomers. It has been shown that two short segments of PrPC containing respectively residues 23–27 and 95–110 are critically important for the efficient binding of PrPC to Aβ oligomers.Citation8,Citation12 In vitro studies show that the PrP23–30 and PrP95–105 peptides efficiently bind to Aβ oligomers and that this interaction is dependent on positively charged residues located in these peptides and also is influenced by the length of the sequence between them.Citation13 It is important to note that PrPC specifically binds to β-conformers but does not interact with Aβ monomer. In contrast, PrPSc polymers interact with Aβ monomers and accelerate Aβ aggregation.Citation14 This suggests that some PrP regions changes their conformations as a result of formation of the polymeric prion, and acquire a capacity of binding Aβ. Specific regions of PrP that are essential for the binding of PrPSc to Aβ remain undefined.
We used a yeast-based system to analyze in vivo the interaction of PrP polymers with Aβ. A number of studies have indicated that S. cerevisiae is a convenient model to study the factors that regulate the misfolding and aggregation of mammalian amyloids.Citation15,Citation16 Various mammalian amyloidogenic proteins, such as α-synuclein, PrP, Aβ and expanded poly-Q region of human Htt, form polymers and/or oligomers in yeast cells that are similar in their biochemical properties to the polymers or oligomers formed in the brains of humans and other animals.Citation6,Citation17-Citation22 In comparison to mammalian systems, Saccharomyces cerevisiae is a simple, cheap and well-studied genetic model that provides significant advantages for the analysis of amyloid interactions. First, the level of protein expression may be adjusted at experimentator’s will in a wide range using regulated promoters. Second, PrP and Aβ aggregates do not kill yeast cells.
Here, we employed a yeast-based assay for the analysis of in vivo interactions between PrP polymers and the 40 amino acid Aβ peptide, and identified the segments of PrP that are crucial for this interaction.
Results
PrP and Aβ fused to fluorescent proteins form amyloid-like polymers in the yeast cytoplasm
To analyze aggregation of mammalian amyloidogenic proteins in yeast, we have expressed Aβ-YFP (or Aβ-GFP) and various fragments of PrP, fused to CFP (constructed as described in Material and Methods) in the yeast cells. Using confocal fluorescence microscopy, we found that Aβ-YFP forms cytologically detectable aggregates in some cells that appear as “dots” or small “clumps” (). All the PrP-CFP fusions formed aggregates of different shapes, but PrP23–231-СFP, PrP28–231-CFP and PrP90–231-СFP preferentially formed dot- or clump-like aggregates, whereas PrP110–231-CFP tended to form tape-like aggregates. We have confirmed by using organelle-specific fluorescent dyes that PrP23–231-GFP and Aβ-GFP aggregates are located in the cytoplasm and do not co-localize with the nucleus, vacuoles, endosome or lipid particles (). To confirm that PrP23–231-CFP, PrP28–231-CFP, PrP90–231-CFP, PrP110–231-CFP and Aβ-YFP proteins form insoluble aggregates in yeast cells, the centrifugation analysis has been performed. After centrifugation, Aβ-YFP and fusions of CFP with PrP derivatives were detected both in the soluble and insoluble fractions, whereas monomeric protein CFP was present only in the soluble fraction ().
Figure 1. Fluorescence microscopy assay of Aβ-YFP and PrP derivatives fused to CFP. Aβ-YFP forms aggregates that appear as “dots” or small “clumps” in some cells and shows diffuse florescence in the other cells. PrP-CFP fusions form aggregates of different shapes: PrP23–231-СFP, PrP28–231-CFP and PrP90–231-СFP preferentially form “dots” or clumps, PrP110–231-CFP tended to form tape-like aggregates. CFP demonstrates diffuse fluorescence.
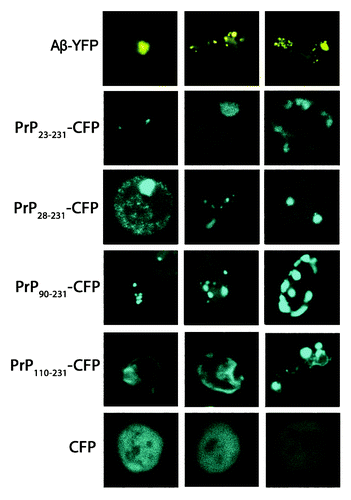
Figure 2. Colocalization of PrP23–231-GFP and Aβ-GFP aggregates with the cell compartments and the lipid particles. The aggregates of PrP23–231-GFP and Aβ-GFP do not co-localize with vacuoles, nucleus, endosomes and lipid particles. The vacuolar membrane (red) was visualized by staining with the lipophilic dye FM4–64. The endosomes (blue) was visualized by staining with Liso-tracker Blue. Nuclear DNA and mtDNA (blue) were stained with DAPI. The staining of lipid particles (black) was achieved by Sudan Black (see Materials and Methods).
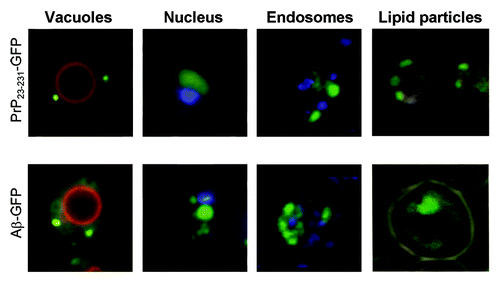
Figure 3. Aβ and full-length PrP or its fragments, fused to fluorescent proteins, exhibit amyloid properties in yeast cells. (A) Analysis of aggregation of Aβ and PrP derivatives fused to YFP and CFP, respectively. Cell lysates were fractionated into soluble (S) and insoluble (I) fractions by centrifugation at 12,000 g, separated on an SDS–PAGE gel, transferred onto a nitrocellulose membrane, and incubated with YFP (CFP)-specific antibody. (B) Analysis of Aβ-YFP and PrP-CFP detergent-resistant polymers by semi-denaturing agarose gel electrophoresis. Cell lysates were treated with 3% sodium lauroyl sarcosinate and analyzed as described using the Ab-13970 antibodies against GFP (Abcam, USA) (see Materials and Methods). (C) Protease K (PK) digestions assay of PrP derivatives and Aβ fused to fluorescent proteins. Samples were digested with 5–100 μg/ml of PK at 37 °C for 1 h. The data presented indicate that all chimeric proteins form aggregates that are highly resistant to proteolysis. The Aβ-YFP protein was detected using the antibodies Ab-13970 against GFP (Abcam, USA) in the sample treated with 40 µg/ml of proteinase K, whereas control GFP completely degraded in the presence of 5 µg/ml proteinase K. Chimeric proteins PrP23–231-CFP, PrP28–231-CFP, PrP90–231-CFP and PrP110–231-CFP, produced resistant fragments after the proteinase K digestion with molecular mass approximately 19–21 kDa. The PrP fragments were detected by the Ab-502604 antibodies against PrP (Abcam, USA). (D) The comparative analysis of PK digestions of PrP23–231-CFP monomers and polymers.
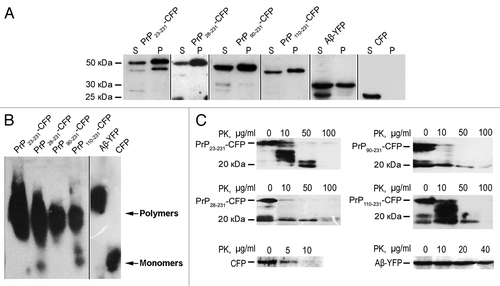
It is known that amyloids exhibit increased resistance to detergents and proteolysis.Citation10,Citation23,Citation24 To determine, whether aggregates of Aβ-YFP and PrP-CFP form detergent-resistant polymers, cell lysates from respective cultures were treated with 3% Sarkosyl (sodium N-lauroylsarcosine) and analyzed by semi-denaturing detergent agarose gel electrophoresis (SDD-AGE).Citation23 Data presented in show that all the chimeric proteins formed detergent-resistant polymers. To analyze the resistance of the Aβ-YFP and PrP-CFP to the digestion by proteinase K, we incubated total cell lysates with various concentrations of proteinase K, separated the samples by SDS-PAGE and probed with an YFP-specific or PrP-specific antibody. The data presented in indicate that all chimeric proteins formed aggregates that are highly resistant to proteolysis. The Aβ-YFP protein was still detected in the sample containing 40 µg/ml of proteinase K, whereas control GFP completely degraded in the presence of 5 µg/ml proteinase K. Increased resistance to PK treatment is the specific feature of the polymeric fraction, as monomers of chimeric proteins were essentially undetectable in the supernatant fraction (not shown), possibly due to their degradation in the absense of protease inhibitors.
It was previously shown that PrP23–231 polymers form in mammalian brains and in yeast cytoplasm a proteinase K-resistant core of approximately 19–21 kDa, which corresponds to amino acids 90 to 231.Citation19,Citation20 Chimeric proteins PrP23–231-CFP, PrP28–231-CFP, PrP90–231-CFP and PrP110–231-CFP, isolated from yeast, produced resistant fragments after the proteinase K digestion with molecular mass approximately 19–21 kDa (). Protease-resistant products reacted to the PrP-specific antibody but were not detected using by the antibody recognizing CFP (data not shown), confirming that they contain only PrP portion of a chimeric protein. Taken together, these results show that Aβ and full-length PrP or its fragments, fused to fluorescent proteins, exhibit amyloid properties in yeast cells.
The PrP28–89 and PrP90–109 segments are involved in the interaction between the PrP polymers and Aβ peptide in yeast
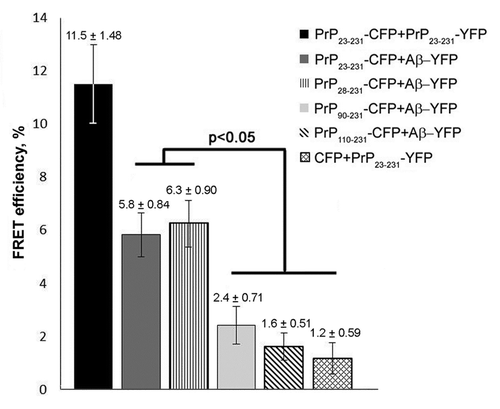
To detect interactions between PrP and Aβ in yeast, we transformed the strain expressing Aβ-YFP with the plasmids expressing PrP23–231-CFP, PrP28–231-CFP, PrP90–231-CFP or PrP110–231-CFP. Resulting transformants co-expressing the fusion genes were analyzed by fluorescent microscopy. The PrP23–231-CFP, PrP28–231-CFP and PrP90–231-CFP polymers co-localized with Aβ-YFP with high frequency (from 75 to 85%) (). The frequency of colocalization of PrP110–231 polymers with Aβ-YFP was approximately five times lower (). The association of PrP aggregates with Aβ was also studied by using a live cell acceptor photobleaching FRET (AΒ FRET) assay. This approach is based on the fact that the energy transfer between the donor (CFP fusion protein) and acceptor (YFP fusion protein) occurs only when the distance between them is not higher than 10 nm.Citation25 Yeast cells co-expressing PrP23–231-CFP, PrP28–231-CFP, PrP90–231-CFP or PrP110–231-CFP with Aβ-YFP were analyzed, while cells co-expressing PrP23–231-CFP with PrP23–231-YFP or PrP23–231-CFP with YFP were used as positive or negative controls, respectively. Photobleaching was performed in the regions of the cells containing the PrP-CFP fluorescent clumps. The greatest FRET efficiency (11.5%) was detected for the cells co-expressing PrP23–231-CFP and PrP23–231-YFP (). FRET efficiency in the cells co-expressing PrP23–231-CFP with Aβ-YFP and PrP28–231-CFP with Aβ-YFP was approximately 6%, whereas the FRET efficiency detected in the cells expressing PrP90–231-CFP and Aβ-YFP was not significantly different from the negative control (). Thus, the PrP fragment comprising amino acids 28 to 89 plays a role in the molecular association of PrP polymers with the Aβ peptide. FRET was not detected in the cells expressing the PrP110–231-CFP and Aβ-YFP proteins. Taken together, these data show that, in addition to the PrP90–109 fragment, the sequence from residues 28 to 89 is also important for the interaction of PrP polymers with Aβ peptide.
Figure 4. Interaction between Aβ and PrP in yeast cells. (A) Analysis of colocalization of PrP23–231-CFP, PrP28–231-CFP, PrP90–231-CFP or PrP110–231-CFP aggregates with Aβ-YFP. Efficient colocalization of PrP23–231-CFP, PrP28–231-CFP and PrP90–231-CFP aggregates with Aβ-YFP was observed. (B) The diagram reflecting the frequencies of colocalization of Aβ-YFP with the PrP23–231-CFP, PrP28–231-CFP, PrP90–231-CFP and PrP110–231-CFP aggregates. The frequencies of colocalization with error bars are indicated.
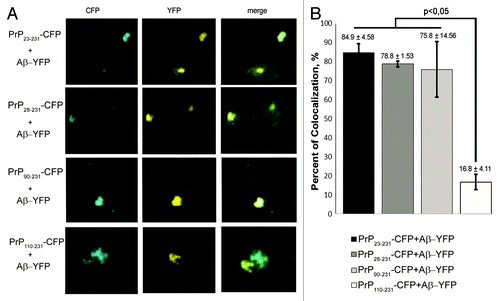
Figure 5. The FRET efficiency between PrP-CFP polymers (donors) and Aβ-YFP (acceptor) that reflects the molecular association of proteins analyzed. The FRET efficiency is shown in percent. Error bars are indicated.
Discussion
Our data confirm the previously demonstrated ability of PrP and Aβ-GFP to form aggregates in yeast,Citation19,Citation26-Citation28 and the similarity in the proteinase K resistance profiles between the PrP aggregates from yeast cells and mammalian PrPSc.Citation19,Citation20 Moreover, we demonstrate that that Aβ-GFP aggregates produced in yeast are also resistant to the digestion by proteinase K. In addition to the previous reported data, were demonstrate that full-length PrP, as well as PrP fragments and Aβ, fused to fluorescent proteins, form detergent-resistant polymers in yeast cells. Therefore, PrP- and Aβ-based aggregates, produced in yeast, exhibit certain amyloid properties. Our results are consistent with recently published data, according to which PrP(90–231) aggregates produced in yeast cells interact with amyloid-specific aptamers.Citation29
We have not detected the colocalization of PrP23–231-CFP or Aβ-YFP with vacuoles, nucleus, endosomes or lipid particles in yeast cells (). Previously, colocalization of Aβ–GFP with lipid particles has been reported by using staining with Nile Red.Citation27 We used Sudan Black for staining lipid particles, because Nile Red fluorescence is detected with both RFP and GFP filters, even in cells not containing the Aβ-GFP protein. Although we cannot exclude that contradictions with the previously published data may be due to differences between the strains or constructs, our data clearly show that colocalization with lipid particles is not the general pattern of Aβ aggregates in yeast. For the first time, our work demonstrates a colocalization between Aβ-YFP and PrP-derived constructs fused to CFP in the yeast cell. Association between PrP23–231 polymers and the Aβ peptide is also proven using FRET, confirming direct physical interaction between these proteins. Our findings demonstrate the suitability of the yeast-based experimental assay for studying interactions between mammalian amyloidogenic proteins. Recently, it was shown that the interaction of Aβ oligomers with PrPC depends on the presence of two short segments in the N-terminal regions of PrP, includes amino acid residues 23–27 and 95–110, respectively.Citation8,Citation12 However, it remained unclear whether or not the same sequences determine the interaction of PrP polymers with Aβ. We have shown that amyloid-like polymers of PrP23–231-CFP efficiently co-localize and physically interact with Aβ-YFP in the yeast cell. The segment 23–27 was dispensable for both co-localization and physical interaction detected by FRET, indicating that it is unlikely to play any role in the interaction between PrP polymers and Aβ. The PrP28–231-CFP and PrP90–231-CFP polymers both co-localized with Aβ-YFP, however FRET detected a physical interaction with Aβ-YFP only for PrP28–231-CFP. One possibility could be that PrP90–231 polymers tend to associate only with aggregated form of Aβ. This decreases the proportion Aβ molecules present in direct contact with PrP, thus decreasing FRET efficiency.
It was demonstrated previously that the positively charged residues in fragments PrP23–27 and PrP90–110 are critically important for the effective binding of PrPC to Aβ oligomers.Citation13 However, the PrP28–89 fragment, which, according our data, regulates the molecular association of PrP polymers with the Aβ peptide, is not enriched with charged amino acids. This suggests that in addition to cationic interactions, other factors influence binding of PrP polymers to Aβ. Further investigation of these factors is made possible by the yeast model developed in our work.
The PrP110–231-CFP polymers, unlike the PrP90–231-CFP, PrP28–231-CFP or PrP23–231-CFP, polymers, co-localized with Aβ-YFP with a low frequency. In this case, it is probable that we observed a non-specific interaction between heterogeneous amyloids, which has previously been described Derkatch et al.Citation30 According to our results, the absence of the PrP90–109 sequence affects the morphology of the PrP polymers and prevents their interaction with Aβ. Based on this result and previously published data,Citation12,Citation13,Citation31 we conclude that the PrP90–109 sequence is required for the interaction of both monomeric and polymeric PrP with Aβ. Additionally, our data show, for the first time, that the PrP28–89 fragment is important for the molecular association between PrP polymers and the Aβ peptide in vivo.
Materials and Methods
Strains and growth conditions
The yeast strain BY4742 (MATα his3Δ1 leu2Δ lys2Δ ura3) (Invitrogen) was employed for this study. Standard yeast genetic techniques, media and cultivation conditions were used;Citation32,Citation33 the yeast cultures were grown at 30 °C. The E. coli strain DH5αCitation34 was grown at 37 °C in Luria-Bertani (LB) broth or on LB agar plates containing ampicillin for plasmid selection.Citation35
Plasmids
The plasmids used in this work are described in . All the plasmids, with the exception of pcDNA3–1-3F4,Citation36 are shuttle vectors that can propagate in S. cerevisiae and E. coli. The vector pRS425 was described previously by Sikorski and Hieter.Citation37 The pSP-YFP and pSP-CFP centromeric plasmids were kindly provided by Dr S Zadorsky (Saint Petersburg State University). The pcDNA3–1-3F4 plasmid contains the mouse Prnp gene modified to express PrP L108M/V111M for immunostaining with the monoclonal antibody 3F4. The plasmid PGPD-GFP(URA3) was described previously by Rubel et al.Citation38 The plasmid PGPD-PrP-GFP(URA3)Citation38 contains the mouse PrP encoding sequence from residue 90 to 231 aa fused in-frame with the GFP ORF and placed under the control of the GPD promoter. The pGPD-YFP(URA3) plasmid was constructed by replacing the 0.7 kb SacII-SacI fragment containing the GFP ORF from PGPD-GFP(URA3) with the 0.7 kb SacII-SacI fragment of yellow fluorescent protein (YFP) obtained from the plasmid pSP-YFP. The pGPD-GFP(LEU2) plasmid was constructed by inserting the 1.4 kb SalI-SacI fragment containing the GPD promoter and the GFP ORF from PGPD-GFP(URA3) into SalI-SacI-digested pRS425. The pGPD-CFP(LEU2) plasmid was constructed by replacing the 0.7 kb SacII-SacI fragment containing the GFP ORF from pGPD-GFP(LEU2) with the 0.7 kb SacII-SacI fragment containing the CFP ORF from pSP-CFP. The plasmids pGPD-PrP23-CFP(LEU2), pGPD-PrP90-CFP(LEU2), pGPD-PrP28-CFP(LEU2) and pGPD-PrP110-CFP(LEU2) were constructed as follows. The BamHI-SacII-flanked sequences of PrP23–231, PrP28–231, PrP90–231 and PrP110–231 were PCR generated from the plasmid pcDNA3–1-3F4 using the FPrP23, FPrP28, FPrP90, FPrP110 and RPrP231 primers listed in . These fragments were inserted into BamHI-SacII-digested pGPD-CFP(LEU2) downstream of PGPD. The pGPD-PrP23-YFP(URA3) plasmid was constructed by inserting the BamHI-SacII fragment containing the PrP23 ORF from pGPD-PrP23-CFP(LEU2) into the BamHI-SacII-digested pGPD-YFP(URA3) plasmid. The PGPD-Aβ-GFP(URA3) plasmid was constructed as follows. The sequence encoding amino acids 1–40 of the human amyloid β peptide (Aβ) was obtained from human brain mRNA by reverse transcription PCR using the FAβ and RAβ primers presented in . The PCR-generated BamHI-SacII fragment was inserted into the PGPD-PrP-GFP(URA3) plasmid digested with BamHI and SacII. The pGPD-Aβ-YFP(URA3) plasmid was constructed by inserting the 0.7 kb SacII-SacI fragment containing the YFP ORF from pSP-YFP into the SacII-SacI-digested PGPD-Aβ-GFP(URA3) plasmid downstream of PGPD.
Table 1. The plasmids obtained in this work
Table 2. Primers used in this study
DNA and protein analysis
Standard procedures were used for DNA isolation and plasmid construction (Sambrook et al., 1989).Citation35 The preparation of the yeast lysates and differential centrifugation were performed according to a described protocol Chernoff et al.Citation39 The fractions were separated by 12% SDS-PAGE. PK-resistant PrP was separated using a 4–20% gradient polyacrylamide gel. SDD-AGE of the PrP-GFP and Aβ-GFP polymers was performed as described Kryndushkin et al.,Citation23 Bagriantsev et al.Citation40 with the modifications. The protein lysates (50–100 μg of total protein) were treated with 3% sodium lauroyl sarcosinate in the sample buffer (25 mM Tris, 200 mM glycine, 5% glycerol, and 0.025% bromophenol blue) for a 7 min at room temperature and run on 1.2% agarose gels. After the electrophoresis, proteins were transferred from gels to Immobilon-P PVDF (GE Healthcare) electrophoretically. Western blotting was performed with the chicken primary monoclonal antibodies Ab-13970 against GFP (Abcam) or with the rabbit primary monoclonal antibodies Ab-502604 against PrP (Abcam). The reactions with the secondary anti-chicken or anti-rabbit antibodies conjugated to horseradish peroxidase and chemiluminescent detection were performed using the ECL system (GE Healthcare). Coomassie staining and Bradford technique were used for normalization of the total protein amount.
Protease digestion assay
All the protease K (PK) digestions were performed in a lysis buffer (20 mm Tris-HCl, 150 mm NaCl, and 1 mm EDTA). The yeast lysates were digested with 5–100 μg/ml of PK at 37 °C for 1 h; the reactions were terminated with 2 mm phenylmethylsulfonyl fluoride.
Fluorescence microscopy
The fluorescence microscopy assays of GFP fusion protein were performed using a Leica DM6000B microscope and Leica QWin Standart 3.2.0. software (Leica Microsystems GmBH). The TCS SP5 confocal laser-scanning microscope and “LAS AF Application Wizard Version 1.7.0” software (Leica Microsystems GmBH) were used for the detailed analysis of aggregates, protein colocalization and live cell acceptor photobleaching FRET (AΒ FRET) experiments. For excitation, we used a 488 nm argon laser for the GFP or GFP fusion proteins and a 594 nm helium-neon laser for FM4–64. The emission filters were 505–515 nm for the GFP or GFP fusion proteins and 615–758 nm for FM4–64. DAPI and Liso-tracker Blue-stained cells were detected using an argon-UV laser with 405 nm excitation and 425–475 nm emission. FM4–64 was detected using a helium-neon laser with 594 nm excitation and 615–758 nm emission. For the FRET experiments, a donor (CFP fusion protein) was co-expressed with an acceptor (YFP fusion protein); for excitation, we used 458 nm and 514 nm argon lasers for the CFP or CFP fusion proteins and YFP or YFP fusion proteins, respectively. The emission filters were 461–510 nm for the CFP or CFP fusion proteins and 518–580 nm for the YFP or YFP fusion proteins. FRET measurements were performed using the FRET acceptor photobleaching program of the Leica confocal software. In the photobleaching procedure, the cells were bleached using a 514 nm laser beam at 100% intensity; the bleach time was 10 s.
For staining with FM4–64 (Invitrogen), mid-log-phase cells were incubated with 8 μM dye (5 mM FM4–64 in dimethyl sulfoxide) for 2 h in liquid selective media. The cells were then washed twice with water and observed for vacuolar staining. The cells were stained with 4,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories) for the visualization of nuclear DNA and mtDNA. The cells were first fixed with 4% formaldehyde for 30 min in culture, washed twice with phosphate-buffered saline (PBS) and permeabilized by incubation in 0.1% PBS Triton X-100 for 10 min. The cells were then washed twice with PBS and incubated for 10 min with VECTASHIELD Mounting Medium (Vector Laboratories) with DAPI (1.5 μg/ml). Liso-tracker Blue (Invitrogen) staining was performed according to the manufacturer’s protocol. The staining of lipid particles in the yeast cells was achieved by Nile Red according to Verstrepen et al.Citation41 or by 0.5% Sudan Black B in ethylene glycol for 60 min according to Thakur et al.Citation42
Statistical analyses
In order to calculate the FRET efficiency, three independent cultures co-expressing Aβ-YFP with PrP23–231-CFP, PrP28–231-CFP, PrP90–231-CFP or PrP110–231-CFP were selected in each cases. The cells that showed both PrP-CFP and Aβ-YFP fluorescence were analyzed. Photobleaching was performed in the regions of the cells containing the PrP-CFP fluorescent clumps. The FRET efficiency (FRETeff) was calculated by software according to the following equation: FRETeff = [Dpost−Dpre]/Dpost, where Dpost is the donor fluorescence after photobleaching and Dpre is the donor fluorescence before photobleaching. We applied the Kruskal-Wallis test with the multiple comparison of z-values for the analysis of the FRET efficiency.
In order to measure frequencies of colocalization between the PrP-CFP polymers and Aβ-YFP, four independent cultures co-expressing Aβ-YFP with PrP23–231-CFP, PrP28–231-CFP, PrP90–231-CFP or PrP110–231-CFP were analyzed in each case. No less than 20 cells containing visible signal of PrP-CFP and Aβ-YFP were analyzed in each culture. The frequencies of colocalization were calculated according to the following equation: [Na/Nt] × 100 where Na – number of PrP-CFP aggregates that co-localize with Aβ-YFP and Nt – total number of PrP-CFP aggregates analyzed. Frequencies were compared by using the Mann-Whitney U-test. Statistical analyses were performed using Statistica v.7.0. (StatSoft Inc., USA) software; P ≤ 0.05 was considered to be statistically significant.
| Abbreviations: | ||
| AD | = | Alzheimer disease |
| Aβ | = | amyloid beta peptide |
| PrPC | = | cellular isoform of prion protein |
| PrPSc | = | prion isoform of prion protein |
| AΒ FRET | = | acceptor photobleaching fluorescence resonance energy transfer |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We are grateful to S Zadorskii for kindly providing pSP-YFP and pSP-CFP plasmids, A Saifitdinova and V Korzhova for helping with FRET experiments, N Degtyareva for critical reading of the manuscript. This work was supported by the program of the Presidium of RAS “Basic sciences—medicine” (to APG), the Ministry of Education and Science of Russian Federation (to YOC and AAR); the grant 1 R03 TW006965 01A1 from Fogarty Center (National Institutes of Health, USA) to YOC and APG; the Russian Federation President grant for young PhDs (MK-1152.2013.4) to AAR; the research grant 1.50.2218.2013 from St. Petersburg State University to YOC. The authors acknowledge the Resource Centers “CHROMAS” (SPbSU) for technical support.
References
- Selkoe DJ, Schenk D. Alzheimer’s disease: molecular understanding predicts amyloid-based therapeutics. Annu Rev Pharmacol Toxicol 2003; 43:545 - 84; http://dx.doi.org/10.1146/annurev.pharmtox.43.100901.140248; PMID: 12415125
- Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat Rev Mol Cell Biol 2007; 8:101 - 12; http://dx.doi.org/10.1038/nrm2101; PMID: 17245412
- Klyubin I, Walsh DM, Lemere CA, Cullen WK, Shankar GM, Betts V, Spooner ET, Jiang L, Anwyl R, Selkoe DJ, et al. Amyloid beta protein immunotherapy neutralizes Abeta oligomers that disrupt synaptic plasticity in vivo.. Nat Med 2005; 11:556 - 61; http://dx.doi.org/10.1038/nm1234; PMID: 15834427
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, et al. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A 1998; 95:6448 - 53; http://dx.doi.org/10.1073/pnas.95.11.6448; PMID: 9600986
- Renner M, Lacor PN, Velasco PT, Xu J, Contractor A, Klein WL, Triller A. Deleterious effects of amyloid beta oligomers acting as an extracellular scaffold for mGluR5. Neuron 2010; 66:739 - 54; http://dx.doi.org/10.1016/j.neuron.2010.04.029; PMID: 20547131
- Wang Y, Meriin AB, Zaarur N, Romanova NV, Chernoff YO, Costello CE, Sherman MY. Abnormal proteins can form aggresome in yeast: aggresome-targeting signals and components of the machinery. FASEB J 2009; 23:451 - 63; http://dx.doi.org/10.1096/fj.08-117614; PMID: 18854435
- Xie L, Helmerhorst E, Taddei K, Plewright B, Van Bronswijk W, Martins R. Alzheimer’s beta-amyloid peptides compete for insulin binding to the insulin receptor. J Neurosci 2002; 22:RC221; PMID: 12006603
- Laurén J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 2009; 457:1128 - 32; http://dx.doi.org/10.1038/nature07761; PMID: 19242475
- Kudo W, Petersen RB, Lee HG. Cellular prion protein and Alzheimer disease: link to oligomeric amyloid-β and neuronal cell death. Prion 2013; 7:114 - 6; http://dx.doi.org/10.4161/pri.22848; PMID: 23154635
- Prusiner SB. Prions. Proc Natl Acad Sci U S A 1998; 95:13363 - 83; http://dx.doi.org/10.1073/pnas.95.23.13363; PMID: 9811807
- Resenberger UK, Harmeier A, Woerner AC, Goodman JL, Müller V, Krishnan R, Vabulas RM, Kretzschmar HA, Lindquist S, Hartl FU, et al. The cellular prion protein mediates neurotoxic signalling of β-sheet-rich conformers independent of prion replication. EMBO J 2011; 30:2057 - 70; http://dx.doi.org/10.1038/emboj.2011.86; PMID: 21441896
- Chen S, Yadav SP, Surewicz WK. Interaction between human prion protein and amyloid-beta (Abeta) oligomers: role OF N-terminal residues. J Biol Chem 2010; 285:26377 - 83; http://dx.doi.org/10.1074/jbc.M110.145516; PMID: 20576610
- Fluharty BR, Biasini E, Stravalaci M, Sclip A, Diomede L, Balducci C, La Vitola P, Messa M, Colombo L, Forloni G, et al. An N-terminal fragment of the prion protein binds to amyloid-β oligomers and inhibits their neurotoxicity in vivo. J Biol Chem 2013; 288:7857 - 66; http://dx.doi.org/10.1074/jbc.M112.423954; PMID: 23362282
- Morales R, Estrada LD, Diaz-Espinoza R, Morales-Scheihing D, Jara MC, Castilla J, Soto C. Molecular cross talk between misfolded proteins in animal models of Alzheimer’s and prion diseases. J Neurosci 2010; 30:4528 - 35; http://dx.doi.org/10.1523/JNEUROSCI.5924-09.2010; PMID: 20357103
- Coughlan CM, Brodsky JL. Use of yeast as a model system to investigate protein conformational diseases. Mol Biotechnol 2005; 30:171 - 80; http://dx.doi.org/10.1385/MB:30:2:171; PMID: 15920289
- Mason RP, Giorgini F. Modeling Huntington disease in yeast: perspectives and future directions. Prion 2011; 5:269 - 76; PMID: 22052350
- Braun RJ, Büttner S, Ring J, Kroemer G, Madeo F. Nervous yeast: modeling neurotoxic cell death. Trends Biochem Sci 2010; 35:135 - 44; http://dx.doi.org/10.1016/j.tibs.2009.10.005; PMID: 19926288
- Gong H, Romanova NV, Allen KD, Chandramowlishwaran P, Gokhale K, Newnam GP, Mieczkowski P, Sherman MY, Chernoff YO. Polyglutamine toxicity is controlled by prion composition and gene dosage in yeast. PLoS Genet 2012; 8:e1002634; http://dx.doi.org/10.1371/journal.pgen.1002634; PMID: 22536159
- Ma J, Lindquist S. De novo generation of a PrPSc-like conformation in living cells. Nat Cell Biol 1999; 1:358 - 61; http://dx.doi.org/10.1038/14053; PMID: 10559963
- Mallik S, Yang W, Norstrom EM, Mastrianni JA. Live cell fluorescence resonance energy transfer predicts an altered molecular association of heterologous PrPSc with PrPC. J Biol Chem 2010; 285:8967 - 75; http://dx.doi.org/10.1074/jbc.M109.058107; PMID: 20086009
- Meriin AB, Zhang X, He X, Newnam GP, Chernoff YO, Sherman MY. Huntington toxicity in yeast model depends on polyglutamine aggregation mediated by a prion-like protein Rnq1. J Cell Biol 2002; 157:997 - 1004; http://dx.doi.org/10.1083/jcb.200112104; PMID: 12058016
- Winderickx J, Delay C, De Vos A, Klinger H, Pellens K, Vanhelmont T, Van Leuven F, Zabrocki P. Protein folding diseases and neurodegeneration: lessons learned from yeast. Biochim Biophys Acta 2008; 1783:1381 - 95; http://dx.doi.org/10.1016/j.bbamcr.2008.01.020; PMID: 18298958
- Kryndushkin DS, Alexandrov IM, Ter-Avanesyan MD, Kushnirov VV. Yeast [PSI+] prion aggregates are formed by small Sup35 polymers fragmented by Hsp104. J Biol Chem 2003; 278:49636 - 43; http://dx.doi.org/10.1074/jbc.M307996200; PMID: 14507919
- Urakov VN, Vishnevskaya AB, Alexandrov IM, Kushnirov VV, Smirnov VN, Ter-Avanesyan MD. Interdependence of amyloid formation in yeast: implications for polyglutamine disorders and biological functions. Prion 2010; 4:45 - 52; http://dx.doi.org/10.4161/pri.4.1.11074; PMID: 20118659
- Ishikawa-Ankerhold HC, Ankerhold R, Drummen GP. Advanced fluorescence microscopy techniques--FRAP, FLIP, FLAP, FRET and FLIM. Molecules 2012; 17:4047 - 132; http://dx.doi.org/10.3390/molecules17044047; PMID: 22469598
- Apodaca J, Kim I, Rao H. Cellular tolerance of prion protein PrP in yeast involves proteolysis and the unfolded protein response. Biochem Biophys Res Commun 2006; 347:319 - 26; http://dx.doi.org/10.1016/j.bbrc.2006.06.078; PMID: 16808901
- Caine J, Sankovich S, Antony H, Waddington L, Macreadie P, Varghese J, Macreadie I. Alzheimer’s Abeta fused to green fluorescent protein induces growth stress and a heat shock response. FEMS Yeast Res 2007; 7:1230 - 6; http://dx.doi.org/10.1111/j.1567-1364.2007.00285.x; PMID: 17662055
- Yang W, Yang H, Tien P. In vitro self-propagation of recombinant PrPSc-like conformation generated in the yeast cytoplasm. FEBS Lett 2006; 580:4231 - 5; http://dx.doi.org/10.1016/j.febslet.2006.06.074; PMID: 16831424
- Mitkevich OV, Kochneva-Pervukhova NV, Surina ER, Benevolensky SV, Kushnirov VV, Ter-Avanesyan MD. DNA aptamers detecting generic amyloid epitopes. Prion 2012; 6:400 - 6; http://dx.doi.org/10.4161/pri.20678; PMID: 22874671
- Derkatch IL, Uptain SM, Outeiro TF, Krishnan R, Lindquist SL, Liebman SW. Effects of Q/N-rich, polyQ, and non-polyQ amyloids on the de novo formation of the [PSI+] prion in yeast and aggregation of Sup35 in vitro.. Proc Natl Acad Sci U S A 2004; 101:12934 - 9; http://dx.doi.org/10.1073/pnas.0404968101; PMID: 15326312
- Lau AL, Yam AY, Michelitsch MM, Wang X, Gao C, Goodson RJ, Shimizu R, Timoteo G, Hall J, Medina-Selby A, et al. Characterization of prion protein (PrP)-derived peptides that discriminate full-length PrPSc from PrPC. Proc Natl Acad Sci U S A 2007; 104:11551 - 6; http://dx.doi.org/10.1073/pnas.0704260104; PMID: 17601775
- Kaiser C, Michaelis S, Mitchell A. Methods in yeast genetics: a Cold Spring Harbor Laboratory course manual.: Plainview, N.Y.: Cold Spring Harbor Laboratory Press, 1994.
- Sherman F, Fink GR, Hancks JB. Methods in yeast genetics. New York: Cold Spring Harbor Laboratory Press, 1986.
- Hanahan D. Techniques for transformation of E. coli. DNA cloning 1985; Glocer, P.M. (ed.), IRL Press, Oxford,1:109-35.
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: a laboratory manual.: Cold Spring Harbor, N.Y.: Cold Spring Harbor Laboratory Press, 1989.
- Narwa R, Harris DA. Prion proteins carrying pathogenic mutations are resistant to phospholipase cleavage of their glycolipid anchors. Biochemistry 1999; 38:8770 - 7; http://dx.doi.org/10.1021/bi990736c; PMID: 10393552
- Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 1989; 122:19 - 27; PMID: 2659436
- Rubel’ AA, Saĭfitdinova AF, Lada AG, Nizhnikov AA, Inge-Vechtomov SG, Galkin AP. [Yeast chaperone Hspl04 regulates gene expression on the posttranscriptional level]. Mol Biol (Mosk) 2008; 42:123 - 30; PMID: 18389629
- Chernoff YO, Uptain SM, Lindquist SL. Analysis of prion factors in yeast. Methods Enzymol 2002; 351:499 - 538; http://dx.doi.org/10.1016/S0076-6879(02)51867-X; PMID: 12073366
- Bagriantsev SN, Kushnirov VV, Liebman SW. Analysis of amyloid aggregates using agarose gel electrophoresis. Methods Enzymol 2006; 412:33 - 48; http://dx.doi.org/10.1016/S0076-6879(06)12003-0; PMID: 17046650
- Verstrepen KJ, Van Laere SD, Vercammen J, Derdelinckx G, Dufour JP, Pretorius IS, Winderickx J, Thevelein JM, Delvaux FR. The Saccharomyces cerevisiae alcohol acetyl transferase Atf1p is localized in lipid particles. Yeast 2004; 21:367 - 77; http://dx.doi.org/10.1002/yea.1100; PMID: 15042596
- Thakur MS, Prapulla SG, Karanth NG. Microscopic Observation of Sudan Black B Staining to monitor Lipid production by Microbes. J Chem Technol Biotechnol 1988; 42:129 - 34; http://dx.doi.org/10.1002/jctb.280420206