
Abstract
The universe of prion and prion-like phenomena has expanded significantly in the past several years. Here, we overview the challenges in classifying this data informatically, given that terms such as “prion-like”, “prion-related” or “prion-forming” do not have a stable meaning in the scientific literature. We examine the spectrum of proteins that have been described in the literature as forming prions, and discuss how “prion” can have a range of meaning, with a strict definition being for demonstration of infection with in vitro-derived recombinant prions. We suggest that although prion/prion-like phenomena can largely be apportioned into a small number of broad groups dependent on the type of transmissibility evidence for them, as new phenomena are discovered in the coming years, a detailed ontological approach might be necessary that allows for subtle definition of different “flavors” of prion / prion-like phenomena.
Originally, the term prion was coined to refer to proteinaceous infectious particles that were shown to cause transmissible spongiform encephalopathies in mammals.Citation1,Citation2 These particles made mainly from mammalian Prion Protein (PrP) were unprecedented, because they do not contain nucleic acids necessary for transmission to individual organisms. Since the first pioneering experiments that discovered prions, the universe of phenomena that are described to be prions or prion-like, has expanded rather a lot. Indeed, designations such as “prion-like” or “prion-related” are used in different ways in different contexts. Given the high profile of prion research, there is always a temptation to use terms like “prion-like” to describe experimental data in order to make the presentation or interpretation of a phenomenon more attractive, or newsworthy. For example, the term “prion-like” can describe domains that have sequences that “look like” prions. In the case of mammalian prions, this might refer to a divergent homolog of the globular Prion Protein domain. Also, sequence tracts that have similar composition to budding-yeast prion determinant domains are often labeled prion-like. In a conceptually different way, “prion-like activity” or “prion-like propagation” may also refer to self-propagating protein aggregates that may not yet meet a stricter prion definition. The latter meaning of “prion-like” is employed in this commentary.
In the simplest sense in the scientific literature, the term “prion” has been used as a name for self-propagating alternative conformations of proteins (i.e., that are propagated sustainably to further protein copies upon binding). In this sense, many amyloids might ultimately be definable as prions. Experimental transmissibility is a proof of such propagation. However, in another sense, the concept of “prion” entails this proven transmissibility. This is either: (1) within the context of diseases, transmissibility into multicellular organisms; (2) within the context of fungal prions, transmissibility into individual fungal cells (unicellular organisms). It is this latter sense of “prion” that we use in this brief discussion. (In a third, most limiting sense, prions are only infectious pathogenic agents, and only PrP prions currently fit this definition, outside of a laboratory context.)
Here, we discuss briefly the issue of how to deal with the diverse prion / prion-like phenomena informatically. To frame this, we refer to a simple three-category classification that we used in the PrionHome database of prion and prion-related sequences.Citation3 These three basic categories of prion/prion-like phenomena are: (1) prion proteins, (2) transcellular prionoids (proteins that demonstrate prion-like propagation between cells within a disease context), and (3) quasi-prions (“quasi-“ meaning “almost”) which are proteins that can underlie prion-like behavior, but do not fit the definitions of (1) or (2).
Prion Proteins
A prion protein is a protein that is known to form prions. A prion is defined here as any unit of sustainable propagation of an altered state of a protein or proteins; the prions are either propagated by infection into individual organisms (as in the case of the mammalian prion protein PrP), or by cytoplasmic/nuclear inheritance to progeny organisms (as for the prions of budding yeast). In the receiving organisms, the prions are propagated through co-option of homologs of the infecting protein(s); these homologs may or may not be transgenic homologs. In the case of PrP, transgenic orthologs from non-laboratory animals are typically introduced into lines of laboratory animals to study the prion propagating ability of these proteins. Obviously, mechanistically, the process of prion transmission for multi-cellular organisms is very different to that for single-celled yeasts.
Such prion proteins have varying degrees of experimental support. They can be divided into two main clear groups of sequences, that are linked to narrower or broader definitions of “prion” (Groups I and II in ). A third group of sequences that are prion-forming as constructs during propagation to yeast mitotic progeny, are included in the Quasi-Prion category (Group III in ). These Groups represent a grading from “proven” to “suspected” prions. They are derived from categories in the PrionHome database (as described in the legend to ), and examples of each Group are listed in the figure.
Figure 1. Classification of prion/prion-like phenomena into broad groups. The prions and quasi-prions can be apportioned into four different broad groups, plus the transcellular prionoids. A narrower prion definition would place Group II in the Quasi-Prions, or conversely, a broader definition would place Group III in the Prions. Group I corresponds to the Type Am* prions in the current PrionHome database release (http://libaio.biol.mcgill.ca/prion), Group II to the Type Am and Ac prions, Group III to the Quasi-prions from Alberti, et al.Citation4 and Group IV to the remaining Quasi-prions. In each box, the protein names and PrionHome database accessions are listed for examples from each category (for prions the name of the prion phenomenon is also given). Complete sets of the categories are available in the database. The database also includes: interactors of prion proteins, paralogs and orthologs of prionogenic proteins, and candidate prion sequences predicted algorithmically,Citation3-Citation5 and tools for visualizing alignments and structures for database entries. Specific experimental details about each prion/prion-like phenomenon are listed in the comments sections of individual database entries.Citation3
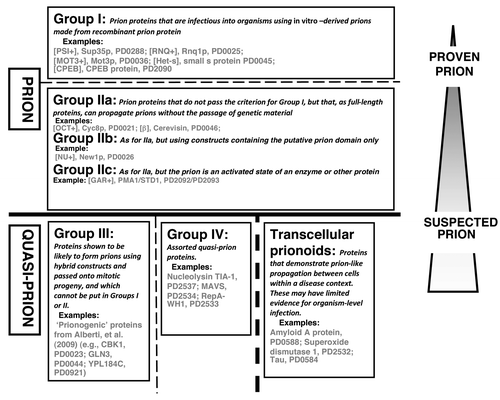
Group I comprises prion proteins that have been demonstrated to be infectious. Several such proteins (e.g., Het-s small s protein, PrP, and Sup35pCitation6-Citation10) have been strongly proven to be able to form prions, in experiments where infectious prion particles formed in vitro from recombinant protein have been introduced artificially into organisms, to initiate sustained prion propagation. Obviously, this proof for prion infectivity is of course very different in terms of experimental design, for single-celled and for multi-cellular organisms. In the case of amyloid-derived prions, this simple criterion of organismal infection is the highest standard of evidence for defining prion-forming proteins. Additional experiments show or robustly confirm the proteinaceous nature of the phenomenon, such as the classic list of criteria derived by Wickner, in the case of fungal prions.Citation11
However, arguably, there might still be problems with such infection experiments; the in vitro prions are derived from recombinant proteins, and, as such, it is not clear how similar they are to genuine in vivo-derived prions, and they are generally less efficient at propagating.Citation12 Furthermore, it has been shown that cofactors of various sorts may be required to promote formation of specific prion strain conformations that are relevant to natural biological prion propagation.Citation10,Citation13,Citation14 Also, despite such successful experiments to infect with in vitro-derived prions, the in vivo context may be complicated by the occurrence of cross-seeding or induction of one prion by another (e.g., the [PIN+]/[RNQ+] prion is required for normal induction of the [PSI+] prionCitation15); i.e., in yeast, any prion is not necessarily a “self-contained” phenomenon, so the relevance of such experiments is not yet completely clear.
The Group I prion proteins include the unusual case of the CPEB protein from Aplysia californica, which contains a glutamine-rich domain that forms amyloids within neurons. This amyloid formation contributes to long-term synaptic facilitation.Citation16,Citation17 However, the amyloid remains inside the neurons, and is not passed on from neuron to neuron, or from organism to organism. CPEB is thus a special case since, unlike the other Group I members, it full-length sequence underlies prion propagation when introduced artificially in budding yeast, even though there is no corresponding prion-forming protein that is native to budding yeastCitation16,Citation17 (although there is a very divergent homolog of CPEB in budding yeast, that actually has a “prion-like” domain, it has nothing to do with the model system that has been set up in yeast to study CPEB prion propagation).
Group II comprises proteins that have not been shown to be infectious using the laboratory protocols described above for infection with recombinant in vitro -derived prions; they do however form prions that are passed onto progeny cells through cytoplasmic (or in some cases, nuclear) inheritance, independent of the passage of genetic material. This has been demonstrated through use of such techniques as cytoduction, or through demonstration of non-Mendelian inheritance. Complementary experiments show or robustly confirm the proteinaceous nature of the phenomenon, such as the classic list of experimental criteria derived by Wickner,Citation11 in the case of fungal prions (reversible curing; overproduction of the protein increasing frequency of prion generation; phenotypic similarity between prion formation and a mutation in the gene making the prion protein). Group II is divided into subgroups IIa, IIb, and IIc (): IIa is for amyloid-based prions that are propagated using the full-length sequences of the prion proteins, whereas IIb is for amyloid-based prions that are propagated using constructs that contain putative prion domains. Group IIb contains the [NU+] prion, which was shown using cytoduction to propagate,Citation18 using a construct of the putative prion domain of the New1p protein replacing the prion domain of the Sup35p protein. Group IIc includes the non-amyloid prions, such as [β], which is the auto-catalyzed active state of the enzyme cerevisin.Citation19 Of course, stricter definition of Prion would place the Group II proteins in the Quasi-Prion category ().
Transcellular Prionoids
The term prionoid was originally used to describe proteins that have prion-like propagation, that sometimes becomes evident on passage between cells.Citation20 Here, a transcellular prionoid is defined as a protein with a prion-like altered state, which demonstrates cell-to-cell prion-like propagation within a disease context. Also, we restrict this definition to proteins from multicellular organisms.
Currently, some of the transcellular prionoids have been demonstrated to cause experimental organismal transmission; however, it is not clear whether other co-factors are necessary for this organismal transmission, or whether the amyloid inoculations are simply “seeding” an on-going process. For example, tauopathy is a neurodegenerative condition linked to the aggregation of Tau protein in the brain, e.g., as in Alzheimer disease, where Tau aggregates are formed in neurofibrillary tangles. This tauopathy can be induced by injection of preparations from the brains of transgenic mice with tauopathy, into transgenic mice expressing human mutant tau.Citation21,Citation22 Also, mechanisms of cell-to-cell propagation of Tau aggregates have been discovered, indicating that Tau is thus a transcellular prionoid.Citation23 However, at the time of completion of this commentary, tauopathy has yet to be induced by injection of in vitro-derived Tau aggregates made from recombinant protein, into an organismal model that does not spontaneously develop tauopathy, meaning that we would not yet classify Tau aggregate propagation as a Group I prion phenomenon.
A potential problem with the transcellular prionoid classification is that, outside of a laboratory context, transcellular propagation also may not be distinguishable from amyloid “seeding” of an already on-going process (as above for organismal transmission).
Quasi-Prions
A quasi-prion is defined as a protein that has some behavior like a prion, but does not fit the definition of either a prion or a transcellular prionoid (). They are assorted protein sequences with specific individual stories.
For example, the quasi-prion mitochondrial antiviral-signaling protein (MAVS) functions in the innate immune response in humans.Citation24 The functional form of MAVS is an amyloid-like aggregate that can co-opt other copies of the protein into the aggregated form. The aggregates are made from a MAVS isoform that plays a role in the assembly of anti-viral signaling complexes on the mitochondrial membrane.Citation24 Also, MAVS aggregates that are formed in vitro can be introduced into cells to propagate the aggregated form to other copies of the MAVS protein, in the absence of viral infection. However, there is currently no evidence that MAVS aggregates can propagate from cell to cell in vivo, or from organism to organism (which would make it a prion, according to the definition above).
The transcellular prionoid sequences could be moved to the Quasi-Prion category, but are kept in a separate category, since they comprise a distinct set of cases in multicellular organisms that demonstrate cell-to-cell propagation relevant to disease processes.
We include in the quasi-prion category, a set of sequences that are “prionogenic” as constructs during propagation to yeast mitotic progeny (these are labeled Group III in ). Some such “prionogenic” proteins have been identified in a large-scale screen for prions that use artificial constructs for some critical experiments.Citation4 Data from fluorescence microscopy, electrophoresis, in vitro assembly assay, and Sup35C prion assay indicated that >20 proteins are likely prionogenic.Citation4 In labeling these sequences as prionogenic, the key experiment was the Sup35C prion assay (in conjunction with evidence for in vivo amyloid formation from the other assays). This assay tests constructs for the ability to produce [PSI+]-like states in yeast cells. The Sup35p protein in yeast functions in translation termination in budding yeast. It contains an N-terminal prion domain, plus a C-terminal globular domain that is essential for protein function. Normally, when full-length Sup35p forms [PSI+] prions, it is taken away from its normal translation termination role, causing stop-codon read-through in the ADE1 reporter gene. In the Sup35C prion assay, to replace the Sup35 prion domain, candidate prion domains were fused to the Sup35p C-terminal protein domain. The candidate prion domains were parts of native yeast protein sequences that were predicted to be prionogenic using an algorithm trained on known prion domain cases. However, these Group III proteins have not been shown more strongly to be prions, using techniques involving the absence of transfer of genetic material, as for New1p (see above).
Concluding Remarks
Classification of prions and prion-like phenomena is an on-going dynamic process. Seizing on one classification does not please every reader, reviewer or editor, as an overview of the literature will indicate. In the broad flexible classification used here, the Groups in are characterized by a type of transmissibility evidence for propagating proteinaceous particles. A narrower definition of prion protein is possible, if we moved Group II to the Quasi-Prion category (). Alternatively, as in a previous version of the PrionHome database,Citation3 we could move Group III to the Prion category. Also, the Transcellular Prionoid category could be made a Group in either the Prion or Quasi-Prion category ().
In the future, when there is a larger corpus of prion data, it will be possible to derive more elaborate hierarchical ontologies of these proteins, in the style of the Gene Ontology (GO).Citation25 The Gene Ontology classifies gene products according to three ontological hierarchical trees (“cellular component”, ‘”molecular function” and “biological process”). In these ontological trees, more general ontological terms such as “nucleic acid-binding” have more specific ‘child’ terms, such as “DNA-“ or “RNA-binding”. For prion/prion-like phenomena, such an ontology would consider: the behaviors of the individual prion/prion-like phenomena both in vivo and in vitro; specific experiments supporting the proteinaceous nature of the phenomena; detail about the constructs used in experiments; and, distinctive characteristics of the sustainable propagation at the molecular level. For example, for yeast prions, such molecular characteristics entail suitably higher relative fragmentation rates that enable infectiousness.Citation26 Another distinguishing characteristic of yeast and other prions may be a lack of the sort of reversibility in amyloid formation that is observed for the functional amyloid Pmel17.Citation27
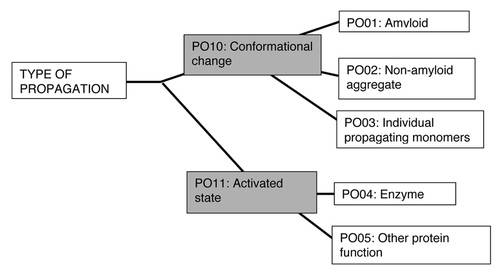
An example of a prion ontological tree is shown, for the “type” of prion propagation (). The ontological terms at each node of such trees could be used to annotate proteins that underlie prion/prion-like phenomena, and give a complete, searchable description of each phenomenon. For example, the yeast protein Rnq1p would be annotated with the “PO01” and “PO10” terms from the tree. As the specific cases of CPEB, Tau, MAVS and [NU+] (discussed above) demonstrate, such a framework of multiple ontological trees would enable more subtle classification of different “flavors” of prion/prion-like phenomena.
Figure 2. An example of a prion ontology hierarchical tree. The “type” of propagation can be classified using a simple hierarchical tree of terms. The most specific terms arise at the leaf nodes of the tree, with more general terms at appropriate internal nodes. Proteins underlying prion or prion-like phenomena can be annotated with terms from multiple such trees. According to this tree, for example, the yeast protein Rnq1p would be annotated with the terms PO01 and PO10.
We hope that this commentary may instigate other investigators to contribute their views on this classification problem, in this journal forum or in others.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Related Research Data
References
- Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science 1982; 216:136 - 44; http://dx.doi.org/10.1126/science.6801762; PMID: 6801762
- Prusiner SB. Prions. Proc Natl Acad Sci U S A 1998; 95:13363 - 83; http://dx.doi.org/10.1073/pnas.95.23.13363; PMID: 9811807
- Harbi D, Parthiban M, Gendoo DM, Ehsani S, Kumar M, Schmitt-Ulms G, Sowdhamini R, Harrison PM. PrionHome: a database of prions and other sequences relevant to prion phenomena. PLoS One 2012; 7:e31785; http://dx.doi.org/10.1371/journal.pone.0031785; PMID: 22363733
- Alberti S, Halfmann R, King O, Kapila A, Lindquist S. A systematic survey identifies prions and illuminates sequence features of prionogenic proteins. Cell 2009; 137:146 - 58; http://dx.doi.org/10.1016/j.cell.2009.02.044; PMID: 19345193
- Harrison LB, Yu Z, Stajich JE, Dietrich FS, Harrison PM. Evolution of budding yeast prion-determinant sequences across diverse fungi. J Mol Biol 2007; 368:273 - 82; http://dx.doi.org/10.1016/j.jmb.2007.01.070; PMID: 17320905
- Maddelein ML, Dos Reis S, Duvezin-Caubet S, Coulary-Salin B, Saupe SJ. Amyloid aggregates of the HET-s prion protein are infectious. Proc Natl Acad Sci U S A 2002; 99:7402 - 7; http://dx.doi.org/10.1073/pnas.072199199; PMID: 12032295
- Stöhr J, Watts JC, Mensinger ZL, Oehler A, Grillo SK, DeArmond SJ, Prusiner SB, Giles K. Purified and synthetic Alzheimer’s amyloid beta (Aβ) prions. Proc Natl Acad Sci U S A 2012; 109:11025 - 30; http://dx.doi.org/10.1073/pnas.1206555109; PMID: 22711819
- Sparrer HE, Santoso A, Szoka FC Jr., Weissman JS. Evidence for the prion hypothesis: induction of the yeast [PSI+] factor by in vitro- converted Sup35 protein. Science 2000; 289:595 - 9; http://dx.doi.org/10.1126/science.289.5479.595; PMID: 10915616
- Legname G, Baskakov IV, Nguyen HO, Riesner D, Cohen FE, DeArmond SJ, Prusiner SB. Synthetic mammalian prions. Science 2004; 305:673 - 6; http://dx.doi.org/10.1126/science.1100195; PMID: 15286374
- Wang F, Zhang Z, Wang X, Li J, Zha L, Yuan CG, Weissmann C, Ma J. Genetic informational RNA is not required for recombinant prion infectivity. J Virol 2012; 86:1874 - 6; http://dx.doi.org/10.1128/JVI.06216-11; PMID: 22090130
- Wickner RB, Edskes HK, Ross ED, Pierce MM, Shewmaker F, Baxa U, Brachmann A. Prions of yeast are genes made of protein: amyloids and enzymes. Cold Spring Harb Symp Quant Biol 2004; 69:489 - 96; http://dx.doi.org/10.1101/sqb.2004.69.489; PMID: 16117685
- Wan W, Wille H, Stöhr J, Baxa U, Prusiner SB, Stubbs G. Degradation of fungal prion HET-s(218-289) induces formation of a generic amyloid fold. Biophys J 2012; 102:2339 - 44; http://dx.doi.org/10.1016/j.bpj.2012.04.011; PMID: 22677387
- Deleault NR, Walsh DJ, Piro JR, Wang F, Wang X, Ma J, Rees JR, Supattapone S. Cofactor molecules maintain infectious conformation and restrict strain properties in purified prions. Proc Natl Acad Sci U S A 2012; 109:E1938 - 46; http://dx.doi.org/10.1073/pnas.1206999109; PMID: 22711839
- Wang F, Wang X, Yuan CG, Ma J. Generating a prion with bacterially expressed recombinant prion protein. Science 2010; 327:1132 - 5; http://dx.doi.org/10.1126/science.1183748; PMID: 20110469
- Derkatch IL, Bradley ME, Hong JY, Liebman SW, Liebman S. Prions affect the appearance of other prions: the story of [PIN(+)]. [PIN+] Cell 2001; 106:171 - 82; http://dx.doi.org/10.1016/S0092-8674(01)00427-5; PMID: 11511345
- Si K, Choi YB, White-Grindley E, Majumdar A, Kandel ER. Aplysia CPEB can form prion-like multimers in sensory neurons that contribute to long-term facilitation. Cell 2010; 140:421 - 35; http://dx.doi.org/10.1016/j.cell.2010.01.008; PMID: 20144764
- Si K, Lindquist S, Kandel ER. A neuronal isoform of the aplysia CPEB has prion-like properties. Cell 2003; 115:879 - 91; http://dx.doi.org/10.1016/S0092-8674(03)01020-1; PMID: 14697205
- Osherovich LZ, Weissman JS. Multiple Gln/Asn-rich prion domains confer susceptibility to induction of the yeast [PSI(+)] prion. Cell 2001; 106:183 - 94; http://dx.doi.org/10.1016/S0092-8674(01)00440-8; PMID: 11511346
- Roberts BT, Wickner RB. Heritable activity: a prion that propagates by covalent autoactivation. Genes Dev 2003; 17:2083 - 7; http://dx.doi.org/10.1101/gad.1115803; PMID: 12923060
- Aguzzi A, Rajendran L. The transcellular spread of cytosolic amyloids, prions, and prionoids. Neuron 2009; 64:783 - 90; http://dx.doi.org/10.1016/j.neuron.2009.12.016; PMID: 20064386
- Sydow A, Mandelkow EM. ‘Prion-like’ propagation of mouse and human tau aggregates in an inducible mouse model of tauopathy. Neurodegener Dis 2010; 7:28 - 31; http://dx.doi.org/10.1159/000283479; PMID: 20160454
- Clavaguera F, Bolmont T, Crowther RA, Abramowski D, Frank S, Probst A, Fraser G, Stalder AK, Beibel M, Staufenbiel M, et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol 2009; 11:909 - 13; http://dx.doi.org/10.1038/ncb1901; PMID: 19503072
- Guo JL, Lee VM. Seeding of normal Tau by pathological Tau conformers drives pathogenesis of Alzheimer-like tangles. J Biol Chem 2011; 286:15317 - 31; http://dx.doi.org/10.1074/jbc.M110.209296; PMID: 21372138
- Hou F, Sun L, Zheng H, Skaug B, Jiang QX, Chen ZJ. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell 2011; 146:448 - 61; http://dx.doi.org/10.1016/j.cell.2011.06.041; PMID: 21782231
- Harris MA, Clark J, Ireland A, Lomax J, Ashburner M, Foulger R, Eilbeck K, Lewis S, Marshall B, Mungall C, et al, Gene Ontology Consortium. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res 2004; 32:D258 - 61; http://dx.doi.org/10.1093/nar/gkh036; PMID: 14681407
- Kushnirov VV, Vishnevskaya AB, Alexandrov IM, Ter-Avanesyan MD. Prion and nonprion amyloids: a comparison inspired by the yeast Sup35 protein. Prion 2007; 1:179 - 84; http://dx.doi.org/10.4161/pri.1.3.4840; PMID: 19164899
- McGlinchey RP, Gruschus JM, Nagy A, Lee JC. Probing fibril dissolution of the repeat domain of a functional amyloid, Pmel17, on the microscopic and residue level. Biochemistry 2011; 50:10567 - 9; http://dx.doi.org/10.1021/bi201578h; PMID: 22092386