
Abstract
Scientific advances in stem cell biology and adult neurogenesis have raised the hope that neurodegenerative disorders could benefit from stem cell-based therapy.
Adult neurogenesis might be part of the physiological regenerative process; however, it might become impaired by the disease's mechanism and therefore contribute to neurodegeneration. In prion disorders this endogenous repair system has rarely been studied. Whether adult neurogenesis plays a role or not in brain repair or in the propagation of prion pathology remains unclear. We have recently investigated the status of adult neural stem cells isolated from prion-infected mice. We were able to show that neural stem cells accumulate and replicate prions thus resulting in an alteration of their neuronal destiny. We also reproduced these results in adult neural stem cells, which were infected in vitro. The fact that endogenous adult neurogenesis could be altered by the accumulation of misfolded prion protein represents another great challenge. Inhibiting prion propagation in these cells would thus help the endogenous neurogenesis to compensate for the injured neuronal system. Moreover, understanding the endogenous modulation of the neurogenesis system would help develop effective neural stem cell-based therapies.
Prion diseases or transmissible spongiform encephalopathies (TSEs) are fatal neurodegenerative disorders, which include Creutzfeldt-Jakob disease in humans, scrapie in sheep and goats, and bovine spongiform encephalopathy in cattle.Citation1 Their origin can be genetic, sporadic, or infectiousCitation1 and currently no treatment is available to prevent widespread neurodegeneration in prion diseases. The incubation period is long before the onset of clinical symptoms and the issue is always fatal. These encephalopathies are called spongiform because of a neuronal vacuolization in the central nervous system (CNS) and transmissible because of the transmission of these diseases. It is possible to inoculate them via intracerebral injections in primates and rodents, which are highly relevant and reliable experimental models. TSE are also characterized by astrocyte proliferation, and especially accumulation of the pathological prion protein PrP scrapie (PrPSc) in the brain.Citation2 This PrPSc corresponds to an abnormal isoform, resulting from the conformational conversion of the cellular host-encoded PrP protein (PrPC) mainly expressed in the CNS.Citation2 According to the prion hypothesis, PrPSc is an infectious particle that can trigger the autocatalytic conversion of the neuronal host-encoded PrPC into PrPSc Citation2 through a still poorly understood misfolding process,Citation2 thus rendering the progression of the disease highly dependent on PrP expression. Several studies have reported an early, severe, and selective loss of a subpopulation of GABAergic interneurons in prion diseases.Citation3 These specific changes in neuronal subsets were identified as a possible reason for some of the clinical symptoms observed in prion diseases.
The diagnosis of these diseases is difficult and often clinicians are faced with a very short therapeutic window after the onset of the first symptoms.Citation4 To date, despite fundamental research's intense focus on prion diseases, no treatment is able to cure or even slow down the disease once symptoms have appeared in animal models or humans. Therapeutic strategies developed so far have only targeted the inhibition of PrPC conversion into PrPSc without treating the lesions that already occurred in the brain. Some investigators have worked on a stem cell-based therapy for prion diseases with limited outcomesCitation5 since grafting fetal neural stem cells (NSCs) from PrP knockout murine embryo did not slow down the progression of the disease. Interestingly, the authors observed an increased number of operational neurons due to the NSC grafting. One of our recent studies consisted in grafting fetal NSCs in prion-infected mice 20 d before the onset of clinical signs. We obtained encouraging results since for the first time in prion diseases we reported an increase in incubation (22.9%) and survival (16%) time.Citation6 In addition to cell replacement, this result could be strengthened by combining cell and gene therapy for a therapeutic effect on the brain.Citation7–9 However, in order to improve the therapeutic effect, many questions still need to be addressed: What is the best time window and location for stem cell grafts? Will grafted NSCs survive? Does prion infection alter the properties of grafted NSCs?
Finding answers to these questions remains essential since a more comprehensive knowledge of the physiopathology of prion diseases, especially for endogenous NSCs, will help the development of new and alternative stem-cell therapies.
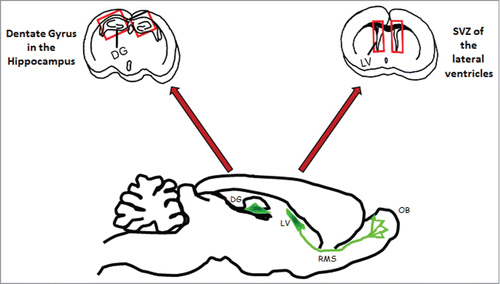
In the past, several studies underlined that tissue-specific stem cells were able to replenish organs by replacing the cells lost to injuries or diseases. For a long time, the mammalian brain was considered the exception to this rule, according to Ramon y Cajal's dogma (1928), postulating that the regenerative capacity of the adult brain was extremely limited. For years, the lack of evidence regarding the existence of endogenous neural stem cells, which could replace cells lost due to injury and disease, was the major argument for the limited regeneration of the adult mammalian CNS. However, in the past decade, several studies have consistently shown that cells with neural stem cell-like properties are found throughout the adult brain. The accumulated evidence has clearly shown that a large number of newborn neurons can be generated from adult NSCs, and integrated into pre-existing neural circuits.Citation10 It is now well known that adult neurogenesis primarily occurs in 2 areas of the mammalian brain (), the dentate gyrus (DG) and subventricular zone (SVZ) of the lateral ventricles.Citation11-15 In the dentate gyrus, newly generated cells within the subgranular layer zone (SGZ) migrate to the granular layer where they differentiate into neuronal cells and extend axonal projections to the CA3 area of Ammon's horn.Citation15 Newborn cells in the anterior part of the SVZ migrate through the rostro-migratory stream (RMS) and integrate the olfactory bulb (OB) where they differentiate into interneurons, granule, and periglomerular neurons.Citation15 Adult neurogenesis in either the SVZ or the DG is also highly sensitive to environmental cues, physiological stimuli, and neuronal activity, suggesting that the tailored addition of new neurons might have an impact on specific neuronal functions. As an example, adult SGZ NSCs are essential for neuron production in the hippocampus and contribute to new memory formation via activity-dependent structural plasticity during our lifetime.Citation16 Even if still debated, recent data also suggested that adult neurogenesis may be found in other regions of the brain such as the amygdala and neocortex.Citation15 In any case, the widespread existence of endogenous neural progenitors represents a new therapeutic pathway for regenerative medicine, either through transplanted NSCs or by manipulating existing ones. Indeed, adult neurogenesis has been suggested as a “neurogenic reserve” that may determine vulnerability to brain dysfunction and neurodegeneration. Since neurogenesis is alterable, its stimulation or potential use of stem cells, recruited endogenously or implanted by transplantation, was suggested as a possible treatment for neurodegenerative disorders.
Figure 1. Neurogenesis areas: frontal sections of the brain of an adult mouse showing in red rectangles: the Dentate Gyrus (DG) in hippocampi; the Subventricular Zone (SVZ) in the Lateral Ventricle (LV). The sagittal sections show in green the neurogenic area in the DG and in the SVZ as well as the rostrary migratory stream (RMS), indicating the migration of neuroblasts into the olfactory bulbs (OB).
The studies on the phenomenon of this de novo neurogenesis within the adult brain have been at the forefront of neuroscience investigations especially in the field of neurodegenerative disorder treatment. However, the balance between neural stem cells activity (proliferation and differentiation) and quiescence is adjusted by environmental physiological and pathological constraints. In brain injury, the increase in adult neurogenesis was shown to be up to 30-fold greater in the rodent brain. This can be associated with a migration of neural precursors toward the injured area.Citation17,18 In certain pathological conditions, new neurons also appear to be generated in local non-neurogenic sites, including the cortex.Citation16 The field of adult neurogenesis is only beginning to be explored for neurodegenerative affections, particularly those involving pathogenic misfolded proteins. Depending on the studies, the results are not always consistent. In humans, for Alzheimer or Huntington diseases, some studies showed the stimulation of the proliferation of neural precursors and that new neurons were generated.Citation19,20 Conversely, in some mouse models of Alzheimer, Huntington, and Parkinson diseases, adult neurogenesis seemed to be altered.Citation21 These distinct neurodegenerative diseases share some common pathogenic and biochemical features, including conversion of a normal cellular protein into a protein with altered conformation and apparent toxic action. Furthermore, several data seemed to indicate that in these proteinopathies, the misfolded proteins implied in the neurodegeneration process are also involved in the regulation of some neurodevelopmental mechanisms.Citation22 In particular, this seems to be the case of Huntingtin in Huntington disease, α-Synuclein in Parkinson disease and APP in Alzheimer disease. In prion diseases, which also involve a pathogenic protein misfolding, the role and status of adult NSCs remain unclear and most fields still need to be explored. Mostly, we do not know if these cells accumulate the misfolded prion protein during the course of the disease. However, it remains unclear whether adult neurogenesis plays a role or not in brain repair or in the propagation of prion pathology.
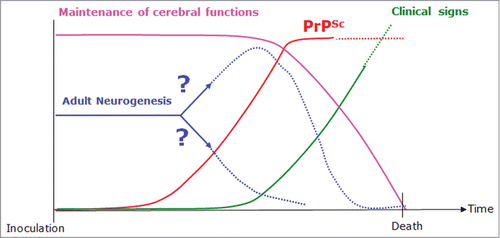
Moreover, one of the fundamental questions in prion disorders addresses the paradox between the duration of the incubation period before the onset of clinical symptoms and the exponential accumulation of the pathological prion protein thought to be toxic.Citation23 One hypothesis could be that adult neurogenesis is stimulated in reaction to the lesions caused by the accumulation of the prion protein in the brain. Even if this stimulation might seem beneficial, the situation is probably more complex for 2 reasons: (1) during neuronal differentiation PrPC expression increases; this PrPC could also be converted into PrPSc, thus promoting the propagation of prions in the brain, (2) PrPC positively regulates the proliferation of neural precursors during developmental and adult neurogenesisCitation24 and seems to be involved in neuronal survival: the pathologic conversion of the protein may therefore modify the neurogenesis and/or contribute to neurodegeneration by altering the function of the prion protein itself.Citation25-27 This self-healing capacity of the adult brain would, however, be insufficient to produce noticeable improvements within the affected neuronal network since only a tiny proportion of cells are successfully integrated and survive. In fact, during the development of the disease, the balance between integrity of the cerebral functions and absence of clinical symptoms could be preserved thanks to the endogenous adult neurogenesis. Then, as a consequence of PrPSc accumulation, a progressive inhibition of the system might occur ().
Figure 2. Diagram summing up our work hypothesis and showing the maintenance of cerebral functions (pink curve), accumulation of PrPSc (red curve), and onset of clinical signs (green curve). The blue curve represents adult neurogenesis which is our subject of interest; we still do not know whether adult neurogenesis is maintained, increased, or decreased. One hypothesis is that during the development of the disease, the balance between the integrity of the cerebral functions and absence of clinical signs could be maintained thanks to endogenous adult neurogenesis. Then, as a consequence of PrPSc accumulation, a progressive inhibition of the system might occur.
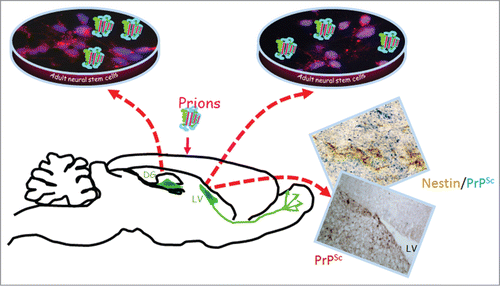
Some of our recent dataCitation28 indicated that PrPSc deposits were densely present in the DG neuronal progenitor area as well as in the neuroblast area surrounding the LVs in the brains of prion-infected mice at the disease's endpoint. Indeed, in mouse brains infected with the ME7 prion strain, immunohistochemical analyses of PrPSc, nestin (NSC marker), and doublecortin (DCX, neuroblast, and immature neuron marker) showed dense PrPSc deposits in the DG and LV neuronal progenitor area as well as in the DCX neuroblasts area surrounding the LV (). By performing a double immunohistochemical analysis of the nestin or DCX markers associated with PrPSc, we clearly validated that PrPSc deposits were located within the nestin and DCX positive cells (, immunohistochemical pictures).
Figure 3. Main results of our recent studies 28: PrPSc accumulates in and around nestin positive cells (pictures on the right). NSC derived from the 2 endogenous neurogenesis areas in prion-infected mice replicate PrPSc (pictures in cell culture dishes at the top). For more information see Relaño-Ginès et al.Citation28
Our objective was to determine whether endogenous NSC and neuronal progenitor cells (NPC) were infected during the progression of the disease. Adult NSC/NPC were therefore isolated from the hippocampus and lateral ventricle of 10 mock and 10 ME7 prion-inoculated mice, 130 d post-infection (dpi). The cells cultivated in free-floating neurosphere conditions were all, as expected, positive for the nestin NSC marker. After 2 sub-passages, we showed that only NSC/NPC derived from ME7-infected mice were positive for PrPSc accumulation (, immunofluorescence pictures). The neuronal differentiation of the infected cells was shown to be compromised since less neuroblasts (DCX marker) and less newborn neurons (betaIII tubulin) were obtained when cells were placed in a neuronal differentiation medium. This was also correlated with an increased amount of astrocytes.
To confirm these results, we also designed ex vivo additional experiments consisting in the infection of adult NSC neurospheres with the ME7 and 22L murine prion strain. In this work we report data on the susceptibility of adult NSC/NPC and the impact this infection might have on their differentiation potential. For this study, NSC/NPC were isolated from LV and DG of adult wild-type mice and PrP-KO mice (from C. Weissmann) and cultivated under free-floating neurosphere conditions. These cells were then infected with 22L and ME7 prions strains. PrPSc was detectable via western blot analysis of cell lysates of hippocampus neurospheres infected with both prion strains (). Control neurospheres incubated with healthy homogenate or even not exposed to ME7/22L prions had no PrPSc at these time points (not shown). PrP-KO control NSC/NPC neurospheres were as expected negative for the presence of PrPSc. PrPSc was also detected in neurospheres derived from lateral ventricles exposed to ME7 and 22L prion strains. PrPSc accumulation was also observed by immunofluorescence analysis in prion infected neurospheres (not shown). The NSC nestin marker was present in most of the dividing NSC cells in both neurospheres ( and ) and disappeared completely after 5 d of differentiation (A). Both infected and non-infected cells were also able to differentiate into astrocytes (GFAP positive cells) and neurons (β III tubulin positive cells) following growth factors withdrawal (B). Yet, as shown with NSC derived from an infected mouse brain,Citation28 the percentage of derived neuroblasts and neurons was significantly lower in infected cells compared with non-infected cells 5 d after differentiation (A–D and ).
Table 1. NSC phénotypes and fates
Figure 4. Adult mouse neurospheres derived from the hippocampus and lateral ventricle of C57Bl6 mice were incubated with 0.1% (p/v) of ME7, 22L, or healthy homogenate during 4 d. Half of the medium was replaced by fresh medium and growth factors (EGF + bFGF) were added every day. When cells reached high density, they were split (1:4 for culture) and (3:4) for analysis by western blot to detect the persistence and/or the de novo production of proteinase K (PK)-resistant PrPSc in infected cultures using a mixture of SAF60, SAF69, SAF70 antibodies. Cells were harvested at 10, 21, 25, 31, 38, and 42 dpi. Western blots show the generation of PrPSc in hippocampus (H) or lateral ventricle (LV) neurospheres from Wild-Type (WT) or PrP Knockout (KO) mice incubated with ME7 and 22L prions. I: WT-H-Healthy inoculum; II: WT-H; III: PrPSc control from ME7 infected brain; IV: PrPSc control from 22L infected brain.
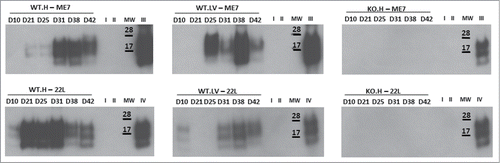
Figure 5. Immunofluorescence analysis of nestin marker in non-infected, ME7 or 22L infected proliferating cells derived from the hippocampus (H) or lateral ventricle (LV) of mice during proliferation conditions (red: nestin; blue: Hoechst nuclei coloration; scale bar, 5 μm).
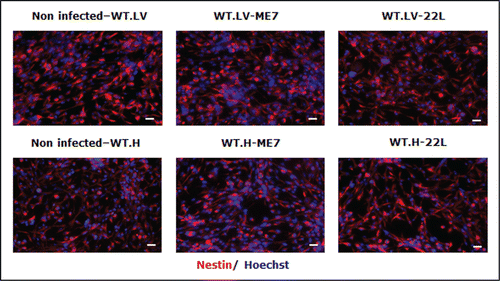
Figure 6. Impairment of the neuronal differentiation of NSC derived from prion infected mice. Immunofluorescence analysis of nestin, DCX, BetaIII-Tubulin, and GFAP astrocytes markers in differentiated NSC derived from the hippocampus (H) or lateral ventricle (LV) that have been infected or not with ME7 or 22L prion strain. (A) red: nestin; green: DCX; (B) green: GFAP; red: BetaIII-Tubulin; blue: Hoechst nuclei coloration; scale bar, 5 μm. (C and D) Quantifications of DCX and β-III-Tubulin positive cells using Cell Profiler software. After differentiation induction, NSC led to significantly more DCX positive cells in non-infected conditions vs. ME7 or 22L infected conditions (Student t test, P < 0.05 is represented by * in the graphic; P < 0.01 is defined by **; and *** corresponds to P < 0.001). The number of neurons also differed significantly between non-infected and infected NSC-derived cells in differentiation conditions. Similar results were obtained in the 3 independent experiments.
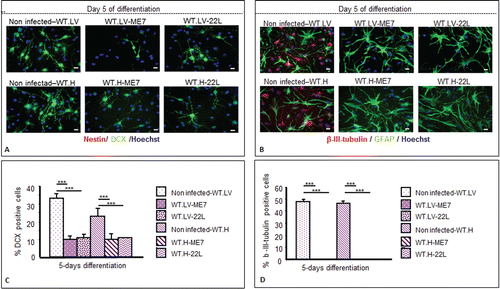
Furthermore, neuronal differentiation impairment () was observed when the infection occurred at the beginning of the differentiation,Citation28 it may underline that some differentiation pathways could be altered when prions replicate in the cells. However, this is not exclusive, as PrPSc might have a potential effect on cell survival or cell proliferation, yet this element needs to be validated by further studies. Concerning other neurodegenerative disorders such as Alzheimer disease, an in vitro study showed that Aβ disrupts the differentiation of NSC.Citation29 A higher proliferation potential was also shown in several models of AD transgenic mice or in AD patients.Citation19,21 Proliferation could therefore be highly relevant since the first observation of prion-infected NSC exhibited a higher proliferation rate compared with non-infected cells (not shown). This could also be coherent with an overactivation of PrPC signaling pathways resulting in cell-cycle modifications.Citation24,30
Although adult neurogenesis might be helpful to the nervous system via regeneration, it might also promote the progression of prion disease. Moreover, the fact that endogenous adult neurogenesis could be altered by the accumulation of misfolded prion proteins represents another great challenge. Inhibiting the misfolding of these pathogenic proteins would therefore allow the endogenous neurogenesis to compensate for the injured neuronal system. Moreover, understanding the endogenous modulation of neural stem cells and the neurogenesis system, could help develop approaches to replenish cellular components lost to injuries and therefore allow the effective development of neural stem cell-based therapies not only for prion disorders but also for other neurodegenerative diseases like Alzheimer or Parkinson diseases. This research is in line with an emerging fundamental field of investigation associating neurodegenerative mechanisms with neurodevelopmental mechanisms. Moreover, the fact that endogenous adult neurogenesis could be regulated within an injured neuronal system represents a promising target for disease-modifying therapies in neurodegenerative diseases.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Bénédicte Clément for proofreading the manuscript.
References
- Collinge J. Prion diseases of humans and animals: their causes and molecular basis. Annu Rev Neurosci 2001; 24:519-50; PMID:11283320; http://dx.doi.org/10.1146/annurev.neuro.24.1.519
- Prusiner SB, Scott MR, DeArmond SJ, Cohen FE. Prion protein biology. Cell 1998; 93:337-48; PMID:9590169; http://dx.doi.org/10.1016/S0092–8674(00)81163–0
- Bouzamondo-Bernstein E, Hopkins SD, Spilman P, Uyehara-Lock J, Deering C, Safar J, Prusiner SB, Ralston HJ 3rd, DeArmond SJ. The neurodegeneration sequence in prion diseases: evidence from functional, morphological and ultrastructural studies of the GABAergic system. J Neuropathol Exp Neurol 2004; 63:882-99; PMID:15330342
- Mallucci G, Collinge J. Rational targeting for prion therapeutics. Nat Rev Neurosci 2005; 6:23-34; PMID:15611724; http://dx.doi.org/10.1038/nrn1584
- Brown KL, Brown J, Ritchie DL, Sales J, Fraser JR. Fetal cell grafts provide long-term protection against scrapie induced neuronal loss. Neuroreport 2001; 12:77-82; PMID:11201096; http://dx.doi.org/10.1097/00001756–200101220–00023
- Relaño-Ginés A, Lehmann S, Bencsik A, Herva ME, Torres JM, Crozet CA. Stem cell therapy extends incubation and survival time in prion-infected mice in a time window-dependant manner. J Infect Dis 2011; 204:1038-45; PMID:21881119; http://dx.doi.org/10.1093/infdis/jir484
- Relaño-Ginés A, Gabelle A, Lehmann S, Milhavet O, Crozet C. Gene and cell therapy for prion diseases. Infect Disord Drug Targets 2009; 9:58-68; PMID:19200016; http://dx.doi.org/10.2174/1871526510909010058
- Crozet C, Lin YL, Mettling C, Mourton-Gilles C, Corbeau P, Lehmann S, Perrier V. Inhibition of PrPSc formation by lentiviral gene transfer of PrP containing dominant negative mutations. J Cell Sci 2004; 117:5591-7; PMID:15494372; http://dx.doi.org/10.1242/jcs.01484
- Toupet K, Compan V, Crozet C, Mourton-Gilles C, Mestre-Francés N, Ibos F, Corbeau P, Verdier JM, Perrier V. Effective gene therapy in a mouse model of prion diseases. PLoS One 2008; 3:e2773; PMID:18648643; http://dx.doi.org/10.1371/journal.pone.0002773
- Cameron HA, McKay RD. Adult neurogenesis produces a large pool of new granule cells in the dentate gyrus. J Comp Neurol 2001; 435:406-17; PMID:11406822; http://dx.doi.org/10.1002/cne.1040
- Taupin P, Gage FH. Adult neurogenesis and neural stem cells of the central nervous system in mammals. J Neurosci Res 2002; 69:745-9; PMID:12205667; http://dx.doi.org/10.1002/jnr.10378
- Alvarez-Buylla A, Garcia-Verdugo JM. Neurogenesis in adult subventricular zone. J Neurosci 2002; 22:629-34; PMID:11826091
- Gage FH. Mammalian neural stem cells. Science 2000; 287:1433-8; PMID:10688783; http://dx.doi.org/10.1126/science.287.5457.1433
- Kempermann G, Wiskott L, Gage FH. Functional significance of adult neurogenesis. Curr Opin Neurobiol 2004; 14:186-91; PMID:15082323; http://dx.doi.org/10.1016/j.conb.2004.03.001
- Ruan L, Lau BW, Wang J, Huang L, Zhuge Q, Wang B, et al. Neurogenesis in neurological and psychiatric diseases and brain injury: From bench to bedside. Prog Neurobiol 2014; 115:116-37; PMID:24384539
- Cayre M, Canoll P, Goldman JE. Cell migration in the normal and pathological postnatal mammalian brain. Prog Neurobiol 2009; 88:41-63; PMID:19428961; http://dx.doi.org/10.1016/j.pneurobio.2009.02.001
- Arvidsson A, Collin T, Kirik D, Kokaia Z, Lindvall O. Neuronal replacement from endogenous precursors in the adult brain after stroke. Nat Med 2002; 8:963-70; PMID:12161747; http://dx.doi.org/10.1038/nm747
- Jin K, Sun Y, Xie L, Peel A, Mao XO, Batteur S, Greenberg DA. Directed migration of neuronal precursors into the ischemic cerebral cortex and striatum. Mol Cell Neurosci 2003; 24:171-89; PMID:14550778; http://dx.doi.org/10.1016/S1044–7431(03)00159–3
- Jin K, Peel AL, Mao XO, Xie L, Cottrell BA, Henshall DC, Greenberg DA. Increased hippocampal neurogenesis in Alzheimer's disease. Proc Natl Acad Sci U S A 2004; 101:343-7; PMID:14660786; http://dx.doi.org/10.1073/pnas.2634794100
- Curtis MA, Penney EB, Pearson AG, van Roon-Mom WM, Butterworth NJ, Dragunow M, Connor B, Faull RL. Increased cell proliferation and neurogenesis in the adult human Huntington's disease brain. Proc Natl Acad Sci U S A 2003; 100:9023-7; PMID:12853570; http://dx.doi.org/10.1073/pnas.1532244100
- Chevallier NL, Soriano S, Kang DE, Masliah E, Hu G, Koo EH. Perturbed neurogenesis in the adult hippocampus associated with presenilin-1 A246E mutation. Am J Pathol 2005; 167:151-9; PMID:15972961; http://dx.doi.org/10.1016/S0002–9440(10)62962–8
- Mehler MF, Gokhan S. Developmental mechanisms in the pathogenesis of neurodegenerative diseases. Prog Neurobiol 2001; 63:337-63; PMID:11115729; http://dx.doi.org/10.1016/S0301–0082(00)00052–6
- Cronier S, Carimalo J, Schaeffer B, Jaumain E, Béringue V, Miquel MC, Laude H, Peyrin JM. Endogenous prion protein conversion is required for prion-induced neuritic alterations and neuronal death. FASEB J 2012; 26:3854-61; PMID:22661006; http://dx.doi.org/10.1096/fj.11–201772
- Steele AD, Emsley JG, Ozdinler PH, Lindquist S, Macklis JD. Prion protein (PrPc) positively regulates neural precursor proliferation during developmental and adult mammalian neurogenesis. Proc Natl Acad Sci U S A 2006; 103:3416-21; PMID:16492732; http://dx.doi.org/10.1073/pnas.0511290103
- Westergard L, Christensen HM, Harris DA. The cellular prion protein (PrP(C)): its physiological function and role in disease. Biochim Biophys Acta 2007; 1772:629-44; PMID:17451912; http://dx.doi.org/10.1016/j.bbadis.2007.02.011
- Pradines E, Hernandez-Rapp J, Villa-Diaz A, Dakowski C, Ardila-Osorio H, Haik S, Schneider B, Launay JM, Kellermann O, Torres JM, et al. Pathogenic prions deviate PrP(C) signaling in neuronal cells and impair A-beta clearance. Cell Death Dis 2013; 4:e456; PMID:23303130; http://dx.doi.org/10.1038/cddis.2012.195
- Simon D, Herva ME, Benitez MJ, Garrido JJ, Rojo AI, Cuadrado A, et al. Dysfunction of the PI3K-Akt-GSK-3 pathway is a common feature in cell culture and in vivo models of prion disease. Neuropathol Appl Neurobiol 2014; 40:311-26; PMID:23741998
- Relaño-Ginès A, Gabelle A, Hamela C, Belondrade M, Casanova D, Mourton-Gilles C, Lehmann S, Crozet C. Prion replication occurs in endogenous adult neural stem cells and alters their neuronal fate: involvement of endogenous neural stem cells in prion diseases. PLoS Pathog 2013; 9:e1003485; PMID:23935493; http://dx.doi.org/10.1371/journal.ppat.1003485
- Haughey NJ, Nath A, Chan SL, Borchard AC, Rao MS, Mattson MP. Disruption of neurogenesis by amyloid beta-peptide, and perturbed neural progenitor cell homeostasis, in models of Alzheimer's disease. J Neurochem 2002; 83:1509-24; PMID:12472904; http://dx.doi.org/10.1046/j.1471–4159.2002.01267.x
- Lee YJ, Baskakov IV. The cellular form of the prion protein is involved in controlling cell cycle dynamics, self-renewal, and the fate of human embryonic stem cell differentiation. J Neurochem 2013; 124:310-22; PMID:22860629; http://dx.doi.org/10.1111/j.1471–4159.2012.07913.x