Abstract
The fruit fly Drosophila melanogaster has been a favored tool for genetic studies for over 100 years and has become an excellent model system to study development, signal transduction, cell biology, immunity, and behavior. The relevance of Drosophila to humans is perhaps best illustrated by the fact that more than 75% of the genes identified in human diseases have counterparts in Drosophila. During the last decade, many fly models of neurodegenerative disorders have contributed to the identification of novel pathways mediating pathogenesis. However, the development of prion disease models in flies has been remarkably challenging. We recently reported a Drosophila model of sporadic prion pathology that shares relevant features with the typical disease in mammals. This new model provides the basis to explore relevant aspects of the biology of the prion protein, such as uncovering the genetic mechanisms regulating prion protein misfolding and prion-induced neurodegeneration, in a dynamic, genetically tractable in vivo system.
Acknowledgements
We would like to thank the support of Joaquin Castilla and Claudio Soto for materials and rich conversations. This work was supported by the John Sealy Memorial Endowment Fund (CON 15431) to D.E.R.-L. and the NIH grant DP2 OD002721-01 to P.F.-F. S.C.-T. was supported by the Kempner postdoctoral fellowships.
Figures and Tables
Figure 1 Hsp70 colonizes membranous domains in response to PrP misfolding. (A) In normal conditions, PrP is synthesized in the ER, modified in the trans-Golgi network (TGN) and secreted through exosomes into the plasma membrane (PM), where it remains attached by a GPI anchor. On the other hand, Hsp70 remains in the cytosol where it contributes to nascent protein folding and protein quality control. (B) Under disease conditions, PrP accumulates in misfolded aggregates (red molecules) in the secretory pathway and the membrane. Hsp70 can detect misfolded PrP and translocates into membranous compartments to interact with PrP. Hsp70 can also be secreted to interact with PrP in lipid rafts.
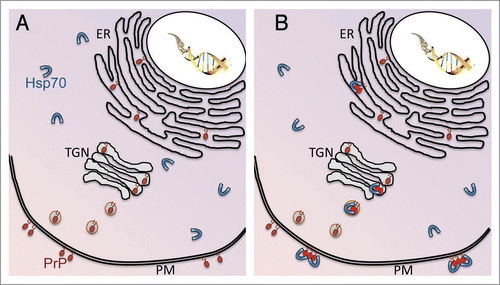
Figure 2 Hsp70 interferes with PrPSc replication in vitro. Brain homogenates from healthy hamsters were inoculated with scrapie-infected brain extracts, and incubated for 72 hours to promote conversion to PrPSc. After incubation, the reactions were digested with PK to visualize PrPSc, except for lane 5 that shows total PrP, and resolved by western blot using the 3F4 antibody. F, Equivalent aliquot of conversion reaction frozen at time zero (no amplification), with (lane 1) and without (lane 5) PK digestion. Incubation and vigorous shaking produced clear amplification of the PK-resistant isoform (lane 2). However, exogenous Hsp70 partially inhibited PrPC conversion (lane 4). This effect is ATP-dependent since addition of ATP further inhibited PrPSc accumulation (lane 3). The result shown is a representative example of four independent experiments and the ratio shown below is normalized against the amplified sample in the absence of Hsp70 (lane 2).

Figure 3 Model for Spontaneous PrP Misfolding and Hsp70 Aactivity. Flies expressing wild type PrP from hamster accumulate a harmless conformer with the biochemical properties of PrPC. Over time, PrP misfolds and converts into a neurotoxic conformation that is insoluble, resistant to denaturing agents, and contains PrPSc epitopes, but is also PK-sensitive (PrP*). This isoform is possibly an intermediary in the metabolism of infectious PrPSc in typical TSE, but it may also exist as an independent pathway. Hsp70 prevents or delays the accumulation of misfolded PrP conformers by either re-folding or tagging them for degradation by components of the Ubiquitin-Proteasome Complex (UPC).
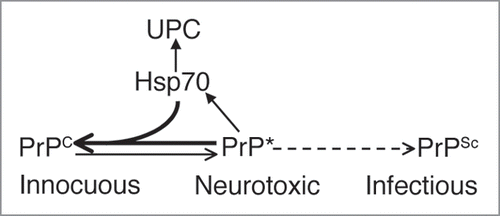
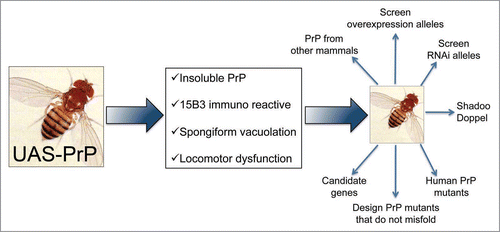
Figure 4 Possible uses for the PrP-expressing flies. Scheme for potential applications of fruit flies expressing mammalian PrP. Flies expressing HaPrP display features relevant in prion disorders, including progressive insolubility, immunoreactivity to the conformational antibody 15B3 that recognizes PrPSc conformers, spongiform vacuolation by day 30 and rapid locomotor dysfunction. These assays can be used as the basis of genetic screens to identify novel genes relevant for PrP misfolding and neurotoxicity, and to undertake complex comparative studies that may result too expensive and time consuming in transgenic mice.