
Abstract
Neurodegenerative diseases are often associated with misfolding and deposition of specific proteins in the nervous system. The prion protein, which is associated with transmissible spongiform encephalopathies (TSEs), is one of them. The normal function of the cellular form of the prion protein (PrPC) is mediated through specific signal transduction pathways and is linked to resistance to oxidative stress, neuronal outgrowth and cell survival. In TSEs, PrPC is converted into an abnormally folded isoform, called PrPSc, that may impair the normal function of the protein and/or generate toxic aggregates. To investigate these molecular events we performed a two-dimensional gel electrophoresis comparison of neuroblastoma N2a cells expressing different amounts of PrPC, and eventually infected with the 22L prion strain. Mass spectrometry and peptide mass fingerprint analysis identified a series of proteins with modified expression. They included the chaperones Grp78/BiP, protein disulfide-isomerase A6, Grp75 and Hsp60 which had an opposite expression upon PrPC expression and PrPSc production. The detection of these proteins was coherent with the idea that protein misfolding plays an important role in TSEs. Other proteins such as calreticulin, tubulin, vimentin or the laminin receptor had their expression modified in infected cells which was reminiscent of previous results. Altogether our data provide molecular information linking PrP expression and misfolding which could be the basis of further therapeutic and pathophysiological research in this field.
Acknowledgements
This work is supported by grants from the “GIS Infections à Prion”, the EU Commission program NEUROPRION (FOOD-CT-2004-506579), the DEFRA project SE2002 and the CNRS. We thank Jacques Grassi and colleagues (CEA Saclay, France) for providing anti-prion antibodies and Patrick Jouin and colleagues from the “Plate-forme de Protéomique Fonctionnelle, IFR3, CNRS-UMR 5203, INSERM-U661” for MALDI-TOF proteomic analysis.
Figures and Tables
Figure 1 Western blot of the cell extracts after PrPC induction & infection. (A) iN2a cells were cultured for three days with increasing amounts of doxycycline. Cells were lysed at 80% of confluence and equal amounts of protein were analyzed by western blotting for PrPC using the antibody SAF32. (B) iN2a cultured for three passages with 200 ng/mL of doxycycline (iN2a+) were infected with 22L brain prion homogenate as detailed in the Materials and Methods section. After passage 3 or 5, control (Ctrl) and infected cells (22L) were analyzed for PrPC and PrPSc expression (PK− and PK+). Infected iN2a+ cells (iN2a+22L) accumulated significant amounts of PrPSc.
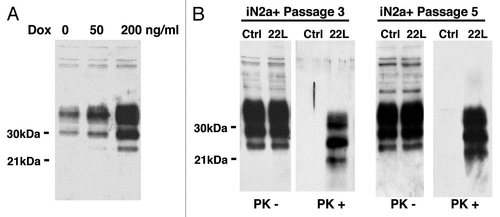
Figure 2 2DE reference gel of iN2a cells. iN2a cell extract were separated using 2DE electrophoresis in a dry strip pH 3–10 for the first dimension, a 12% SDS-PAGE for the second dimension and silver stained. The identification of proteins, noted with their ID number (Suppl. Tables 1 and 2), was performed by peptide mass fingerprints after trypsin digestion and MALDI-TOF on Coomassie or silver stained spots.

Figure 3 Distribution of the identified proteins per functional blocks (A) and cellular origin (B). (References in Suppl. Table 2.)
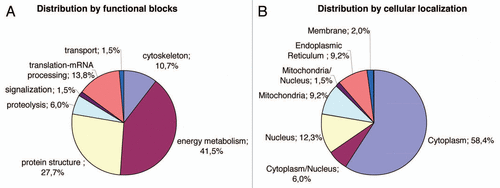
Figure 4 Illustration of protein differential expression. (A) Regions of 2DE silver stained gel of the different cell types illustrating the variation of calreticulin, ATP synthase, lactate dehydrogenase and voltage-dependent anion-selective channel protein. (B) Western blot detection after 1D gel electrophoresis of PDIA6, tubulin beta and vimentin in iN2a, iN2a+ and iN2a+22L. Numbers in parenthesis refer to protein ID numbers (Suppl. Tables 1 and 3).
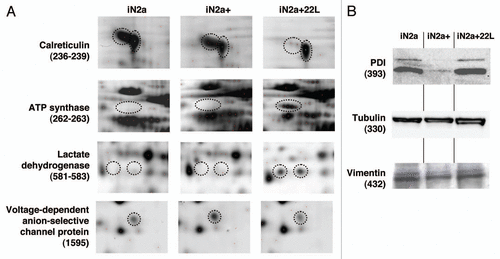
Table 1 Table of identified proteins (spot ID, gene, accession number from UniprotKB database, name and functional group) sorted in eight groups (A-H) based on the fold expression ratio iN2a+/iN2a and iN2a+22L/iN2a+