Abstract
Neurodegenerative diseases are caused by proteinaceous aggregates, usually consisting of misfolded proteins which are often typified by a high proportion of β-sheets, which accumulate in the Central Nervous System. These diseases, including Morbus Alzheimer, Parkinson disease and Transmissible Spongiform Encephalopathies (TSEs) also termed prion disorders, afflict a substantial proportion of the human population and as such the etiology and pathogenesis of these diseases has been the focus of mounting research. Although many of these diseases arise from genetic mutations or are sporadic in nature, the possible horizontal transmissibility of neurodegenerative diseases poses a great threat to population health. In this article we discuss recent studies which suggest that the “non-transmissible” status bestowed upon Alzheimer and Parkinson diseases may need to be revised as these diseases have been successfully induced through tissue transplants. Furthermore, we highlight the importance of investigating the “natural” mechanism of prion transmission including peroral and perenteral transmission, proposed routes of gastrointestinal uptake and neuroinvasion of ingested infectious prion proteins. We examine the multitude of factors which may influence oral transmissibility and discuss the zoonotic threats which Chronic Wasting Disease (CWD), Bovine Spongiform Encephalopathy (BSE) and Scrapie may pose resulting in vCJD or related disorders. In addition, we suggest that the 37 kDa/67 kDa laminin receptor on the cell surface of enterocytes, a major cell population in the intestine, may play an important role in the intestinal pathophysiology of alimentary prion infections.
Acknowledgements
We thank the National Research Foundation (NRF), Republic of South Africa for financial support.
Figures and Tables
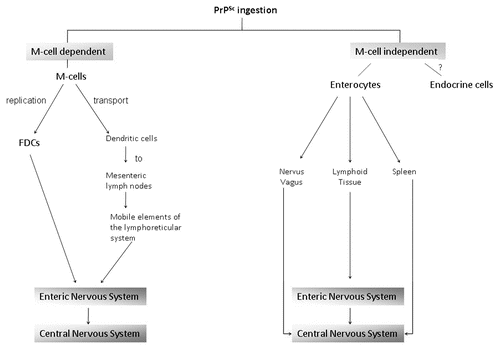
Figure 1 Proposed routes of gastrointestinal entry of ingested infectious prions (PrPSc) as well as possible pathways of amplification and transport to the central nervous system.