Abstract
Genetic Creutzfeldt-Jakob disease (gCJD) is caused by a range of mutations in the prion protein gene (PRNP) and account for approximately 10-15% of overall human prion diseases worldwide. They are different with disease onset, disease duration, clinical signs and diagnostic findings. Here we reported a 71 year-old female with an E196K mutation in one PRNP allele, while the codon 129 was a methionine homozygous genotype. The patient started with non-specific symptoms, but displayed rapidly progressive disturbances of speech, memory, cognitive and physical movement. No periodic activity was recorded at electroencephalography (EEG) during the entire disease course. Retrospective investigation of her family members did not reveal similar neurological disorders. Total clinical course was about seven months.
Introduction
Human prion diseases or Creutzfeldt-Jakob disease (CJD), are a group of neurodegenerative diseases of central nerve system (CNS). The pathological agent of the diseases, termed as PrPSc, is believed to be an abnormal conformational conversion isoform of PrPC.Citation1 Sporadic CJD cases have no unambiguous cause and represent about 80–85% of human prion diseases.Citation2 Genetic CJD is related with prion protein gene (PRNP) mutation on chromosome 20 and account for approximately 10–15% of human prion cases.Citation3 More than 55 mutations in PRNP have been confirmed to be associated or directly linked with the development of genetic human prion diseases noted.Citation4 While about 1% cases are iatrogenically acquiredCitation4 and more than 250 vCJD cases have been reported worldwide following contamination by agent of bovine spongiform encephalopathy.Citation5
The national surveillance for CJD has been conducted in mainland of China since 2005. Different genetic CJD (gCJD), fatal familial insomnia (FFI) and Gerstmann-Sträussler-Scheinker syndrome (GSS) cases have been identified.Citation6–Citation8 Here we reported a 71-year-old Chinese woman suffered from gCJD of E196K mutation in PRNP, who started with non-specific symptoms without family history of the similar neurological disorders.
Case Presentation
A Chinese female, who was a 71-year-old retired worker, complained to have glossolalia and decreased spontaneous speech for three weeks. The symptoms deteriorated sharply and one week later she failed to communicate with her family members. About ten days before admission, she was noted to have hypomnesia and listlessness and was unable to recognize her husband and son. Meanwhile, she developed dysphagia. One week before entering the hospital, her physical movement weakened gradually and her limbs were stiff sometimes. On admission, she was able to walk with support by others, but became bedridden subsequently. Two weeks later, she developed dysuria and hallucination at night. Her body temperature and blood pressure were at the normal range during the course of hospitalization. Neurological examinations showed severe apathy, mutism, flexion and adduction of forearms, flexion of carpal joint and fingers, generalized hyperreflexia of the limbs and bilateral Babinski signs. Myoclonus was not observed throughout the clinical course. Her general situation worsened progressively and she discharged from hospital two months later. At the terminus of the clinical course at home, she appeared severe akinetic mutism, coma and cachexia. She died in hometown four months after discharge. Total clinical course was about seven months. Retrospective investigation of her family members did not reveal similar neurological disorders. No brain autopsy was obtained and her family members refused to do further genetic examination.
During hospitalization, no periodic activity was recorded in electroencephalography (EEG). Magnetic resonance imaging (MRI) revealed abnormal hyperintense lesions on T1 and T2-weighted image within the bilateral corona radiate and centrum semiovale. On diffusion-weighted MRI, hyperintense lesions were noted in the right medulla oblongata and revealed asymmetrical cortical atrophy. Biochemistry and cell count of cerebrospinal fluid (CSF) was normal.
About 30 days after the onset of clinical symptoms, CSF was collected by lumbar puncture. CSF 14-3-3 protein was detected with western blot according to the protocol described elsewhere with minor modifications.Citation9 Briefly, 20 µl CSF sample was separated by 12% SDS-PAGE and electronically transformed onto nitrocellulose membrane. Blots were incubated in 1:1,000 diluted 14-3-3 polyclonal antibodies (Santa Cruz, CA) and further incubated in 1:5,000 diluted HRP-conjugated goat anti-Rabbit IgG (PerkinElmer, Germany). Immunoreactive bands were visualized by ECL method (PerkinElmer, Germany). No 14-3-3 specific signal was observed by western blots in CSF. Her genomic DNA was extracted from peripheral blood leukocytes and the coding region of PRNP was amplified by a PCR protocol,Citation10 with human PRNP specific forward primer (5′-GGC AAA CCT TGG ATG CTG G-3′) and reversed primer (5′-CCC ACT ATC AGG AAG ATG AGG-3′). Direct sequencing of the PCR products with an automatic genetic analyzer (ABI3130XL) revealed a methionine homozygous genotype at codon 129 of PRNP. A missense mutation G to A at the position of nt 586 in one PRNP allele was identified, leading to a change from a glutamic acid (E) to lysine (K) mutation at residue 196 (). No other mutation was observed in the rest of the PRNP gene.
Discussion
In this report, we have described the first Chinese E196K gCJD case. The onset of disease is rapid, with decreased spontaneous speech and glossolalia, which has been initially considered and treated as cerebral infarct. Along with the clinical progression, other neurological manifestations appear, i.e., memory and cognitive disturbances, dyskinesia, which let the physician assume other neurological disorders. In fact, uncertain and diversiform initial symptoms of CJD often cause the clinician to make a misdiagnosis, which accounts for many gCJDs, especially for those without family histories. The progression of the clinical presentations of this patient is seven months.
Additional to this case, ten other E196K gCJD cases have been reported since identifying the first one in 2000,Citation11 including the six German patients described more recently in reference Citation12. The main clinical characteristics of the cases are comparably summarized in . The patients were clinically presented with nonspecific symptoms at disease onset including speech problems, dementia, depression and sleep disturbance. During the process of disease, motor signs, akinetic mutism, severe apathy were developed gradually. Myoclonus seems to be frequently observed. The disease related family history is notified in five cases, including the recently reported four German cases belonging to two different families Citation12 and the first French case whose mother and brother died with dementia and motor impairment without definite diagnosis.Citation9 Other six cases seem lacking of definite family history.Citation13–Citation15 Onset ages of E196K gCJD range from 64–80 year-old (median: 71 year-old), which are obviously later than that of most other gCJD cases. The durations of the disease range from 2–18 months (median: seven months), which are somewhat shorter than that of some other kinds of genetic human prion diseases.Citation16,Citation17 The onset ages and clinical durations of the cases having family history are similar as that of the ones without family history. All cases lack of typical periodic sharp wave complexes (PSWC) in EEG, even though some cases only had sharp wave but without periodicity. Except the patient reported in this study, all patients show positive CSF 14-3-3 protein, possibly related with relatively earlier sampling of CSF (30 days after onset) in this case. Ten patients are homozygote at codon 129, including 7 of 129 (M/M) and 3 of 129 (V/V). Only one case is heterozygote in 129 (M/V). Neuropathologic features of E196K gCJD are merely obtained from four German cases, showing meningeal vessels and amyloid angiopathy with less demyelination in the white matter consistent with small vessel disease.Citation10
From the three-dimensional analysis of the human PrP, it showed that the E196K mutation is located sequentially adjacent to the third alpha helix of PrP. The mutation at the point could consequentially eliminate the salt bridge that establishes a link between the first and third alpha helix, between E196 and R156. In a result, this could induce a reduced stability of the K196 PrP variant which would promote the accumulation of mutated PrP protein in brain.Citation18
In conclusion, we have reported here the first Chinese E196K gCJD case that is the eleventh one worldwide. Most E196K gCJD cases are sporadic without clear family history. The onset ages of the disease seem to be later and durations are relatively short. The clinical manifestations are somehow like that of sCJD, but without typical PSWC in EEG. Further attentions are needed in order to avoid of misdiagnosis, especially for the elder cases having CJD-like symptoms but without typical EEG abnormality and family history.
Figures and Tables
Figure 1 Graphic presentation of the sequencing analysis of PRNP. The presence of mutation is confirmed by direct sequencing of PRNP after PCR amplification. It shows a G to A heterozygous transition at codon 196 in one PRNP allele, leading to an exchange from Glu (E) to Lys (K). The arrow above the curves indicates the position where both G and A are present. The sequences of nucleotides and the encoding amino acids are illustrated at the bottom of graph.
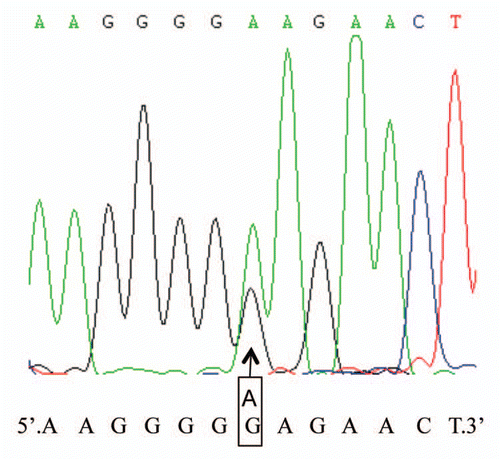
Table 1 Clinical, electroencephalographic, 14-3-3 and genetic features of patients with the PRNP E196K mutation
Acknowledgments
This work was supported by Chinese National Natural Science Foundation Grants 30771914 and 30800975, Institution Technique R&D Grant (2008EG150300), National Basic Research Program of China (973 Program) (2007CB310505), China Mega-Project for Infectious Disease (2009ZX10004-101) and the SKLID Development Grant (2008SKILD102, 2011SKILD211 and 2011SKILD204).
References
- Prusiner SB. Prions. Proc Natl Acad Sci USA 1998; 95:13363 - 13383
- Brandel JP. Clinical aspects of human spongiform encephalopathies, with the exception of iatrogenic forms. Biomed Pharmacother 1999; 53:14 - 18
- Capellari S, Strammiello R, Saverioni D, Kretzschmar H, Parchi P. Genetic Creutzfeldt-Jakob disease and fatal familial insomnia: insights into phenotypic variability and disease pathogenesis. Acta Neuropathol 2011; 121:21 - 37
- Hilton DA. Pathogenesis and prevalence of variant Creutzfeldt-Jakob disease. J Pathol 2006; 208:134 - 141
- The National Creutzfeldt-Jakob Disease Surveillance Unit (NCJDSU). www.cjd.ed.ac.uk/index.htm
- Shi Q, Gao C, Zhou W, Zhang BY, Tian C, Chen JM, et al. Human prion disease with a T188K mutation in Chinese: a case report. Cases J 2009; 29:7820
- Gao C, Shi Q, Zhou W, Tian C, Jiang HY, Han BY, et al. The first Chinese case of Creutzfeldt-Jakob disease with mutation of E200K in PRNP. Biomed Environ Sci 2010; 23:158 - 160
- Shi XH, Han J, Zhang J, Shi Q, Chen JM, Xia SL, et al. Clinical, histopathological and genetic studies in a family with fatal familial insomnia. Infect Genet Evol 2010; 10:292 - 297
- Cuadrado-Corrales N, Jiménez-Huete A, Albo C, Hortigüela R, Vega L, Cerrato L, et al. Impact of the clinical context on the 14-3-3 test for the diagnosis of sporadic CJD. BMC Neurol 2006; 6:25
- Shi Q, Gao C, Zhou W, Zhang BY, Chen JM, Tian C, et al. Surveillance for Creutzfeldt-Jakob disease in China from 2006 to 2007. BMC Public Health 2008; 8:360
- Peoc'h K, Manivet P, Beaudry P, Attane F, Besson G, Hannequin D, et al. Identification of three novel mutations (E196K, V203I, E211Q) in the prion protein gene (PRNP) in inherited prion diseases with Creutzfeldt-Jakob disease phenotype. Hum Mutat 2000; 15:482
- Schelzke G, Eigenbrod S, Romero C, Varges D, Breithaupt M, Taratuto AL, et al. Genetic prion disease with codon 196 PRNP mutation: clinical and pathological findings. Neurobiol Aging 2011; 32:1 - 9; http://dx.doi.org/10.1016/j.neurobiolaging.2010.11.023
- Tumani H, Windi O, Kretzschmar HA, Ludolph AC. Clinically atypical CJD: diagnostic relevance of cerebrospinal fluid markers and molecular genetic analysis?. Dtsch Med Wochenschr 2002; 127:318 - 320
- Clerici F, Elia A, Girotti F, Contri P, Mariani C, Tagliavini F, et al. Atypical presentation of Creutzfeldt-Jakob disease: the first Italian case associated with E196K mutation in the PRNP gene. J Neurol Sci 2008; 275:145 - 147
- Béjot Y, Osseby GV, Caillier M, Moreau T, Laplanche JL, Giroud M. Rare E196K mutation in the PRNP gene of a patient exhibiting behavioral abnormalities. Clin Neurol Neurosurg 2010; 112:244 - 247
- Ye J, Han J, Shi Q, Zhang BY, Wang GR, Tian C, et al. Human prion disease with a G114V mutation and epidemiological studies in a Chinese family: a case series. J Med Case Reports 2008; 2:331
- Iwasaki Y, Kizawa M, Hori N, Kitamoto T, Sobue G. A case of Gerstmann-Sträussler-Scheinker syndrome with the P105L prion protein gene mutation presenting with ataxia and extrapyramidal signs without spastic paraparesis. Clin Neurol Neurosurg 2009; 111:606 - 609
- Guest WC, Cashman NR, Plotkin SS. Electrostatics in the stability and misfolding of the prion protein: salt bridges, self energy and solvation. Biochem Cell Biol 2010; 88:371 - 381