Abstract
Prion diseases are infectious conformational diseases. Despite the determination of many native prion protein (PrP) structures and in vitro production of infectious prions from recombinant PrP the structural background of PrP conversion remains the largest unsolved problem. The aggregated state of PrPSc makes it inaccessible to high resolution techniques, therefore indirect methods have to be used to investigate the conversion process. We engineered disulfide bridges into the structured domain of PrP in order to determine the secondary structure elements that remain conserved upon conversion. Rather surprisingly, introduction of disulfides into each or both of the subdomains B1-H1-B2 and H2-H3 of the C-terminal globular domain retained the robust ability to convert into fibrils with increased content of β-structure, indistinguishable from the wild-type PrP. On the other hand disulfide bridges tethering the two subdomains completely prevented conversion, while their reduction reversed their conversion ability. The same conversion propensity was replicated also in prion infected cell lines. Experiments with combinations of engineered cysteine residues further support that domain swapping, centered on the B2-H2 loop, previously associated to species barrier, leads to PrP swapped dimers as the building block of prion fibrils.
Acknowledgments
The authors acknowledge the financial support from the Slovenian Research Agency (I.H.B., R.J.), EN-FIST Centre of Excellence (R.J.), and the sixth framework EU project, TSEUR.
Figures and Tables
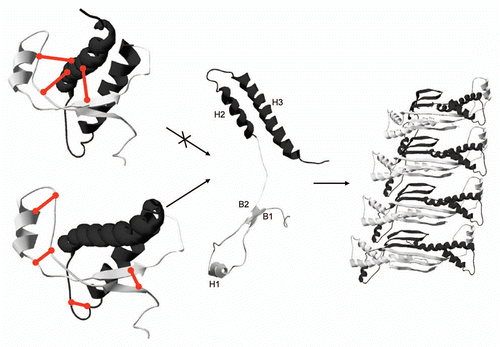
Figure 1 Mapping of PrP conversion by disulfide tethers. Disulfides engineered within the globular domain of PrP have different effects on its ability to convert into fibrils. Disulfide tethers are schematically represented as straight connectors on mouse PrP structure (1XYX).Citation22 All disulfides (left top), which tether on one side subdomain B1-H1-B2 (gray) and on the other subdomain H2-H3 (black) prevent conversion, while PrP variants with single or even double disulfide tethers within each or both of the two subdomains retain the ability to convert into fibrils (left bottom). Results suggest that the secondary structure of each of the two subdomains is conserved during conversion, which can be accomplished by separation of subdomains (middle) followed by domain swapping. Domain swapped PrP dimer thus represents the building block of fibrils and a template for the annealing of the disordered N-terminal part into β-structure. Monomers within a swapped dimer are shown in gray and black (right).