
Abstract
The suggested role of cellular prion protein (PrPC) in mediating the toxic effects of oligomeric amyloid β peptide (Aβ) in Alzheimer disease (AD) is controversial. To address the hypothesis that variable PrPC expression is involved in AD pathogenesis, we analyzed PrPC expression in the frontal and temporal cortices and hippocampus of individuals with no cognitive impairment (NCI), amnestic mild cognitive impairment (aMCI), mild AD (mAD), and AD. We found that PrPC expression in all brain regions was not significantly altered among the various patient groups. In addition, PrPC levels in all groups did not correlate with expression of methionine (M) or valine (V) at codon 129 of the PrP gene, a polymorphism that has been linked in some studies to increased risk for AD, and which occurs in close proximity to the proposed binding region for the oligomeric Aβ peptide. Our results indicate that, if PrPC is involved in mediating the toxic effects of the oligomeric Aβ peptide, these effects occur independently of steady state levels of PrP or the codon 129 polymorphism.
Acknowledgments
This study was supported by grants 5P30 AG028383 and 1P01AI077774-015261 to GCT from the National Institute on Aging (NIA), and the National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health (NIH); AG027219 to SWS from the NIA, NIH, and by the Sanders Brown Center on Aging, University of Kentucky Medical Center, Lexington, KY.
Figures and Tables
Figure 1 Representative restriction fragment length polymorphism (RFLP) analyses. Lanes 1 and 2, brain extracts from Tg(HuPrP-M129)6816+/− mice expressing human PrP-M129; lanes 3 and 4, brain extracts from and Tg(HuPrP-V129)7823+/− mice expressing human PrP-V129; lanes 5 and 6, equal mixture of brain extracts from transgenic mice expressing human PrP-M129 and transgenic mice expressing human PrP-V129; lanes 7–16, five representative human samples. Odd numbers, undigested PCR samples; even numbers, digested PCR samples. (A) PCR amplified samples treated with restriction enzyme NspI, which cleaves PRNP encoding methionine at codon 129. Methionine carriers produce a 363 bp fragment; valine carriers produce a 438 bp fragment. (B) PCR samples treated with MaeII, which cleaves PRNP encoding valine at codon 129. Valine carriers produce a 359 bp fragment.
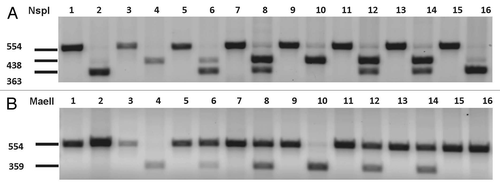
Figure 2 Rostrocaudal analysis of PrPC levels in NCI and AD patients. For all samples, levels of total PrPC and actin were measured by densitometric scanning of western blots. Each PrP value was normalized to its actin value. Normalized PrPC levels were compared among the frontal (F) and temporal (T) cortices and hippocampus (H) in NCI and AD groups. Error bars represent standard deviations from the means.
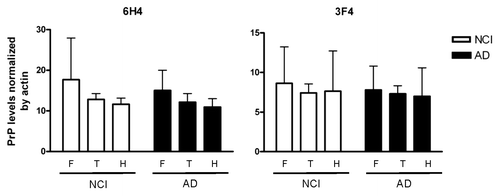
Figure 3 Levels of PrPC in different brain regions of patients with differing levels of cognitive impairment. Representative western blot analyses of brain homogenates from NCI, aMCI, mAD and AD patients are shown. Anti-PrP 6H4 (A–C) and 3F4 (D–F) antibodies were used to compare the levels of PrPC among the groups. Actin levels are shown below the 6H4 and 3F4 immunoblots. NCI: lanes 1–4. aMCI: 5–8. mAD: 9–12. AD: 13–16. Three brain regions including the frontal (A, D, G and J), temporal (B, E, H and K) cortices and hippocampus (C, F, I and L) were analyzed. Molecular markers indicated are 53, 36, 28 and 19 kDa. Graphs in (G–L) include analyses of all samples from each group. For each sample, levels of total PrPC were measured by densitometry. For each sample, levels of actin were also assessed. Each PrP value was normalized to its actin value. Means and standard deviations were calculated for each patient group and expressed as the percent value relative to the NCI group.
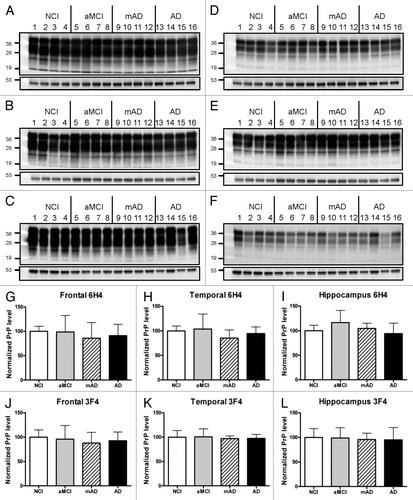
Figure 4 Unaltered levels of unglycosylated PrPC in AD patients compared to NCI individuals. Levels of unglycosylated PrPC (∼molecular weight 25–30 kDa) were measured by densitometric analysis of western blots. Means and standard deviations were calculated for samples from each patient group. y-axis values are arbitrary densitometric units. Asterisks indicate p > 0.05.
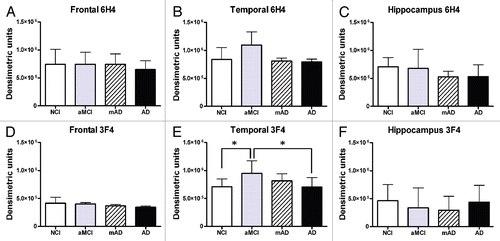
Figure 5 Levels of PrPC do not correlate with human PrP 129 polymorphism in AD patients. Levels of PrPC were measured by densitometry. For each sample, levels of actin were also assessed. Each PrP value was normalized to its actin value. These values were compared among individuals with various PRNP genotypes (M/M, M/V, V/V). (A–C) represent the data analyzed with 6H4; (D–F) represent the data analyzed with 3F4. Three brain regions including the frontal (A and D) and temporal (B and E) cortices and hippocampus (C and F) were analyzed.
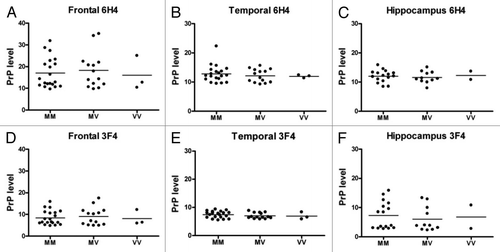
Table 1 General demographics of subjects
Table 2 Braak scores by clinical diagnosis
Table 3 CERAD classification by clinical diagnosis
Table 4 Codon 129 PRNP allele frequency and genotype of subjects