Abstract
The subacute spongiform encephalopathies are prion diseases characterized by acute and rapid neurodegeneration that lead to the death of the patient within months to a few years. The epidemiology of CJD is complicated and the frequency in Mexico is unknown. We aim to describe the cases of prion disease in Mexico. Consecutive patients who met the diagnostic criteria by the WHO were enrolled. We describe 26 patients with clinical manifestations, imaging and laboratory studies compatible with prion disease. The mean age at onset was 52 years old. The main clinical manifestations were cognitive alterations (69%) followed by extrapyramidal movements (50%), abnormal cerebellar function (46%), behavioral alterations (46%), myoclonus (46%), and mood depression (23%), among other features. Half of the patients progressed rapidly to a state of akinetic mutism (53%). Only 2 (7.6%) patients had a family history of a similar disease. Time interval between onset and diagnosis varied between 71 days to 24 months, with a median of 6 months. The classical bilateral basal ganglia hyperintensities were present in the very early stage of the disease. Protein 14-3-3 immuneassay in the CSF was positive in all measured cases. Bilateral basal ganglia hyperintensities was the most important early finding, while protein 14-3-3 was a late finding and the results were usually obtained after the patient was discharged. Around 1.5 cases of CJD cases per year are reported in our country. When suspected, MRI can support the diagnosis earlier than other studies.
Background
The subacute spongiform encephalopathies are prion diseases characterized by acute and rapid neurodegeneration that leads to the death of the patient within months to a few years. They comprise a group of four diseases in humans: Kuru, Creutzfeldt-Jacob disease (CJD), Gerstmann-Sträussler Syndrome and Fatal Familiar Insomnia, with CJD being the most common human prion disease at the moment.Citation1,Citation2
CJD is characterized by a long period of incubation followed by a rapid progressive course. It affects both men and women, showing the greatest frequency among patients aged 45–75 years old. The main clinical symptoms consist of mood changes, psychoaffective and sleep disorders, rapidly progressive dementia, hyperesthesia, hyperreflexia, muscular fasciculations, tremors, myoclonus (abrupt jerking movements of muscle groups or the entire limbs), visual ailments, and other pyramidal and extrapyramidal signs.Citation1,Citation3 Tests used to orient the clinical diagnosis of CJD are the 14-3-3 protein in the cerebrospinal fluid (CSF 14-3-3), electroencephalogram (EEG) and magnetic resonance imaging (MRI).Citation1–Citation3 No treatment for CJD exists at the moment, only management of symptoms.Citation3
The sporadic CJD (sCJD) form is responsible for 85–90% of CJD cases. Several hypothetical mechanism for the origin and spread of sporadic CJD have been proposed, including exposure to infected meat, or the appearance of spontaneous somatic mutations to yield an infectious protein agent de novo. On the other hand, the familial form features a dominant inheritance pattern.Citation1,Citation3 This form is caused by somatic mutations that occur in the prionic gene (PRNP) located on the short arm of chromosome 20 at codon 102.Citation1 Lastly, iatrogenic transmission has also been described.
The epidemiology of CJD is very complicated. It has been recognized worldwide, at rates of 0.25 to 2 cases per million per year. In Mexico, there are only 3 previous reports of CJD casesCitation3–Citation5 among other reasons, due to limited knowledge concerning this disease on the part of the medical staff, which causes a lack of notification of cases, and an under-registration of the disease.Citation1 On the other hand, there are no centers or laboratories of microbiology and genetics where tests to support the diagnosis of the disease can be conducted.Citation1 This is the case of most developing countries. However, the following cases demonstrate that the clinical course is surprisingly characteristic, and the findings in the MRI and EEG can accurately support the diagnosis if the clinical suspicion is strong. We aim to provide evidence that prion disease is more common than supposed. With this, we hope to increase the awareness of the clinical features of CJD.
Results
Seven cases of suspected CJD cases were admitted to our Institution from 1999 to 2011. Demographic and clinical characteristics are individually described and summarized on . shows the MRI images and the characteristic EEGs of these cases. In addition, we included two videos of cases 1 and 7. summarizes our findings with other the three reports of Mexican CJD cases found in the literature. Searches were performed through Pubmed using the terms “Creutzfeld-Jakob Diseae”, “prion disease”, “Protein 14-3-3”, “Mexico”, “Mexican”. To our knowledge, only 26 cases of CJFD have been reported in our country.
Case 1.
A 59-year old right-handed woman developed a rapidly progressive mental disease and involuntary movements. Her family history was relevant for an uncle with an unknown rapidly progressive mental disease. She was born in Pachuca, Estado de México and lived in Mexico City. She did not eat red meat regularly, nor did travel frequently, and was not exposed animals or toxins. She had been diagnosed with type-2 diabetes mellitus 8 years earlier and developed peripheral neuropathy and retinopathy. She was admitted for a 2 month history of feeling anxious, irritable and forgetful, with loss of the circadian rhythm of sleep. One month later she developed involuntary myoclonic movements of the upper extremities. Her gait became unstable and her speech dysarthric. She lost 37 pounds (17 kilos). Bilateral tremor in upper extremities emerged along with severe incoordination. She began to tumble and crash with objects. One week before her admission she had a severe fall and she was brought to the emergency department. At her arrival she was sleepy. When woken-up, her language was non-fluent and very soft. Her facial expression was lost. She was able to understand and follow orders, to name objects and repeat sentences, but she was easily distracted and her thought was slow. Her left arm was clumsy. At that time her MMSE was 26 points. Cranial nerve examination showed slow eye movements. Muscle strength was normal. Tone was generalized increased. Deep tendon reflexes were 2+ in the left side and 3+ in the right side. Plantar reflexes were normal. She had bilateral tremor that was worse in the left side. The finger-to-nose test was abnormal, as were the other coordination movements. Gait was ataxic. Frontal release signs were present. Five days after her hospitalization her MMSE declined to 11 points. Blood tests were normal, including thyroid function tests, VDRL and serology for EBV. CSF analysis showed glucose of 74 mg/dL, proteins of 52 mg/dL and no cells. PCR for herpes simplex virus, varicella zoster virus and JC virus were negative. CSF gram stain, Ziehl-Nilsen stain and the cryptococcal antigen were negative. Antineoplasic antibodies (anti-Ri and anti-Yo) in CSF were also negative. The brain MRI showed bilateral hiperintesities in caudate and putamen nucleus (). Protein 14-3-3 was positive (4,200 UI) in CSF. She died one month after being discharged.
Case 2.
A 60-year old man developed dementia rapidly. His mother died of a similar disease at an unknown age. He was born and lived in Poza Rica, Veracruz. He had finished elementary school and was a businessman. He denied toxic exposures or drug use. He was admitted to the hospital for a 3 month history of altered mental status, dizziness and weakness in the lower extremities. One month later he presented spontaneous involuntary movements during sleep. He was prescribed carbamazepine without improvement. Two weeks later he developed aggressive behavior that alternated with a state of perplexity and indifference to the surroundings. He also developed speech alterations and visual hallucinations. There was an involuntary weight loss of 20 kg. He was admitted to our hospital for further study. Upon admission he was awake, but he did not follow simple commands. He was disoriented in time and place. His language was soft, non-fluent and dysarthric. MMS was of 14 points. His muscle strength was decreased, worse in lower than in the upper extremities. He had a left Babinski sign. The cerebellar function was severely impaired, he had altered finger-to-nose test and impaired coordinating movements worse in the left side. He showed normal liver function tests, normal thyroid tests and negative anti-thyroid antibodies. Vitamin B12 levels (1,297 pg/ml) and serum calcium (9.7 mg/dL) were within normal range. HIV was negative. A brain CT only showed cortical atrophy. The EEG showed generalized spikes (). In the CSF analysis cell count was negative as were the cultures and stains, but there was a marked increase in the 14-3-3 protein. He was discharged and did not have further follow-up.
Case 3.
A 66-years old woman developed a rapidly dementing syndrome. She was a widower and housekeeper. She lived in Michoacán. She was exposed to wood smoke for over 20 years. She denied other toxic exposures or drug use. She had contact with hens and dogs. Her left kidney was removed for unknown reasons 20 years before, and two years ago she was diganosed with type-2 diabetes. She was admitted for headache, dizziness, slowness in thinking and speech that began one year earlier and worsened three months before addmission. At her arrival she was alert but her attention span was impaired, and she was disoriented. Her speech was fluent but incongruent. The cranial nerve examination, muscle strength and reflexes were normal. Babinski signs were absent. The sensory examination was normal. There were no abnormal movements. She had frontal release signs. The finger-nose test was abnormal in both sides, as were the rapid alternating movements. She had severe ataxia and her gait was unstable. One month later she had difficulty for making eye contact, her speech became non-fluent and dysarthric. She had eye wondering and was unable to follow simple commands. VDRL was negative, vitamin B12 serum levels were inbeteween ranges and she had normal liver and thyroid function tests. Anti-Hu antibodies were indetermiated, but anti-Yo antibodies were negative. CSF analysis showed a leucocyte count of 2 cells/mm3, and PCR for tuberculosis was negative. Her MRI showed hyperintensity of basal ganglia suggestive of CJD. Her EEG showed diffuse slow activity with periodics sharp waves. She worsened in weeks, as she was unable to follow commands and was mute and acinetic. She was discharged home.
Case 4.
A male of 57 years of age came for altered mental function and involuntary movements. He lived in Morelos, finished elementary school and worked as a gardener. He had a cousin that died of a neurological disease that consisted of encephalopathy and seizures. He smoked tobacco occasionally, but had no other toxic exposures or drug use. He began 3 months before his admission with involuntary movements on his left upper arm and slow speech. He had a normal CT scan. One month later he was unable to communicate or walk. He had trouble swallowing, generalized rigidity and slowness in his movements. His coordination was worse on the left side. His laboratory studies were normal. CSF analysis was normal. The MRI showed bilateral hyperintensities in the basal ganglia (). His EEG showed slow activity with triphasic waves in the right midtemporal region. He was discharged and no further follow-up.
Case 5.
A 60-years old male had rapid dementia. He was married, completed elementary school and worked as a policeman. He was from Coacalco, Estado de México, and lived in Mexico City. He smoked (Tobacco index of 21) and drank alcohol heavily every other week since age 22. He began two months before admission with dizziness, sleepiness and severe headache that woke him at night. He was irritable, very emotional and complained of auditory hallucinations. One month later, involuntary myoclonic movements developed on the right arm, as well as tremor in his left arm. At his arrival he showed impaired concentration and difficulties in performing mental tasks, and he had impaired short span memory. His thought was slow and his speech was dysarthric. He had severe difficulties in finding objects in front of him. His cranial nerve examination was relevant for vertical nistagmus and bilateral sensoryneural hypoacusia. Mucular tone was increased in the left side. He had spontaneous myclonic movements in the upper and lower extremities. He had ataxia and altered rapid alternating movements. While walking he tended to fall to the left side. His laboratory studies showed marked elevation of TSH and he was prescribed with levothyroxine. Liver function tests and electrolytes were normal. Vitamin B12 levels were normal (>15,000). Nor VRDL or anti-thyroid antibodies were done. He had a first EEG that showed diffuse slow activity and a second EEG performed one week later showed periodic sharp waves (). His CSF analysis showed proteins of 60 mg/dL, and severe elevation of 14-3-3 protein. He died one month later.
Case 6.
A 50 year-old woman developed dementia rapidly. She was divorced and worked as a secretary. She lived in Mexico City. She smoked and drank alcohol occasionally. She began one year before with muscular weakness, muscular atrophy and involuntary movements in the upper extremities. She began falling to the right side and experiencing dizziness and upper extremity rigidity. At her arrival she was inattentive, disoriented and unable to follow simple commands. Her speech was non-fluent. Cranial nerve examination was relevant for vertical nistagmus and left XI nerve paralysis. Muscle strength was 3/5 in upper extremities and 0/5 in the lower extremities. Deep tendon reflexes were 3+ in the upper and lower extremities. She had a fixed flexed posture on both arms above the torax and of both legs above the hip. There was severe spasticity. There were muscle fasciculations in the upper extremities. She was unable to perform the finger-nose test and her movements were uncoordinated on both sides. Frontal release reflexes were present. Cognitive functions declined within three months. The patient was mute and did not make eye contact with other persons. Liver function tests were normal, seric sodium was 128 mEq/L, VDRL, HIV and PPD were negative. Thyroid function was normal, as it was the vitamin B12 levels, and the antithyroid, anti-Yo and anti-borrellia antibodies. Seric heavy metal levels were normal. Her CSF showed increased proteins of 64 mg/dL with normal glucose and no cells. VHS PCR, cryptococcus antigen and black ink stain were negative. Her MRI showed hyperintensity in the basal ganglia, and the EEG showed triphasic waves (). The 14-3-3 protein on the CSF was positive. She died two months later.
Case 7.
A 38-year old woman born in Guerrero came because a rapidly developing psychiatric illness associated with involuntary movements. She began 11 months earlier with abnormal behavior, easy crying and somnolence. She was very irritable and fought constantly with her husband. She was diagnosed with mood depression and started on SSRIs. She began having episodes of unexplained aggression and haloperidol and quetiapine were prescribed. Five months later she developed visual hallucination and insomnia. She presented rapid and non-rhythmic coarse involuntary movements of the lower and upper extremities. She began walking on her toes and her balance was impaired. She stumbled and fell frequently. She began presenting continuous involuntary movements that were also present at night. Soon after she was unable to walk. Ten months later she was admitted to the hospital. Her MMSS was 12/30 and her speech was dysarthric. Muscle strength was 4/5, but deep tendon reflexes and muscle tone were normal. Hoffmann and Tromner signs were present, but she did not show Babinski signs. She had severe ataxia and severe alterations on the finger-to-nose test and of the rapid alternating movements. She had myoclonus of the upper and lower limbs. Frontal release signs were not present. She had normal laboratory results including thyroid function tests, and antinuclear antibodies. HIV and VDRL were negative. Heavy metal screening for aluminum, copper, arsenic, nickel, zinc and lead were negative. The CSF examination showed glucose of 58 mg/dL, proteins of 38 mg/dL and cero cells. Oligoclonal bands were negative. The MRI showed bilateral hypeintense images in the basal ganglia () with enlarged ventricles. The EEG showed slow background activity, poor gradient, poor reactivity to stimuli and sparse triphasic waves on the left frontotemporal region. Protein 14-3-3 was positive in the CSF. Akinetic mutism developed and she was discharged.
Discussion
The occurrence and significance of spongiform encephalopathies in Mexico is unknown. Four cases were reported from year 1996 to 2001,Citation4,Citation5 and 18 more cases were detected between year 2000 and 2005.Citation3 We are adding 7 new cases identified in our Institution from January 1999 to February 2010, counting for a total of 26 patients (). According to the World Health Organization criteria,Citation1 six (23%) were classified as definite, ten (38%) as probable and ten (38%) possible. The overall incidence is relatively low when compared to the reports from other countries.
The mean age at onset was 52 years old, slightly younger than what it is reported in the literature,Citation1,Citation3,Citation4 where mean age for sporadic CJD is 60 to 69 years old. The main clinical manifestations were cognitive alterations (69%), followed by extrapyramidal movements (50%), abnormal cerebellar function (46%), behavioral alterations (46%), myoclonus (46%) and mood depression (23%), among other features (). Half of the patients progressed rapidly to a state of akinetic mutism (53%). Only two (7.6%) patients had a family history of a similar disease. More than one third (39%) of the patients were born and lived in Mexico City, and three of the four centers that reported the cases were also in this location.
Time interval between onset and diagnosis varied between 71 days to 24 months, with a median of 6 months. According to the Slow Virus Infection Research Committee stages of the diseaseCitation6 most of the patients were seen on stages I (nervousness, insomnia, headache, dizziness, mood depression or anxiety) before admission to our facilities, 19 (65%) were seen at stage II (occurrence of a distinct neurological syndrome with a prominent involvement of cortical functions and hallucinations), and only 10 (38%) arrived at a later stage (severe reduced movement or complete akinetic mutism).
CSF findings were irrelevant in most cases, as only mild proteinorraquia was present in some. However, for the past decade, new diagnostic tests have shown to be extremely helpful. Albeit virtually all patients with CJD have abnormal EEG findings, most of them are non-specific at the early stages.Citation2,Citation11 However, the importance of performing an EEG relies on the fact that abnormalities only rarely occur in patients with rapidly progressive dementia of other etiologies such as Alzheimer's disease, vascular dementia or Lewy body disease.Citation2,Citation11
Increased signal intensity in caudate nucleus, putamen and parietal or occipital cortical areas in fluid-attenuated inversion recovery (FLAIR) and diffusion-weighted (DW) MRI sequences were seen in all of the cases who had an MRI (), and in almost half of the patients of the other series. This finding was the earliest and most consistent positive result. We think that the distinguishing features of the MRI, along with a typical clinical course, can be used as the basis of the diagnosis when 14-3-3 protein in the CSF is not available. As described before, DWI abnormalities have a higher sensitivity (92%) and specificity (93–98%), and may be detected as early as 3 weeks of symptom duration.Citation7–Citation9
Detection of protein 14-3-3 in the CSF has been the most promising surrogate marker for prion disease in the last years, for which a specificity of 84% and a sensitivity of 94% have been reported in reference Citation5, Citation10 and Citation11. Although CSF protein 14-3-3 was positive in most cases, the result often came back after the patient was discharged. Brain biopsy may be diagnostic of CJD, however, this procedure can be recommended only if some other potentially treatable disease remains to be excluded.
The patient of case 7 has several interesting findings. First, she was relatively young for sporadic (sCJD), as she was 38 years old at admission. The median age at death for sCJD is 68 years old, and for vCJD is 28 years old. She also displayed prominent psychiatric and behavioral symptoms at onset, and the overall duration of illness was 11 months. Although these features are more consistent with the variant form of the disease (vCJD) than with sCJD, in which duration of the disease is usually shorter, between 4 and 5 months and dementia and neurologic signs are present earlier or at the same time of the psychiatric manifestations, against vCJD is the presence of periodic sharp waves on the electroencephalogram and no evidence for suspecting iatrogenic or acquired disease.
No occupational activities were more prevalent and the origin of the sporadic disease could not be eluded from this study. We found two patients who had a family history of a similar disease. It is generally considered that between 10% and 15% of CJD cases have a family history of the disease.Citation3
Our study has several deficiencies. First, we do not perform biopsies or autopsies in all of our patients. Second, PrP genotyping is available in other countries and is useful for genotyping the patients, unfortunately it is not performed in our country or similar developing countries. Even when CSF 14-3-3 protein determination may be carried out in some medical centers as special studies, it was not possible to perform it in all the patients. Lastly, we do not have the time of the duration of the disease in many cases, mostly because it was considered that a close follow-up in a specialized center far from their local environment would not change the clinical course and would only add the inconvenience of the trip.
Non-the less, we consider that this study reveal important information about CJD in our country. There is a tendency to underestimate the real frequency of the disease, and it is possible that it is more common than it appears, but less diagnosed due to technical and clinical deficiencies.
Methods
An observational, descriptive and transversal study was conducted. Information was recorded relating to cases of CJD in the Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán in Mexico City between January 1999 and February 2011. Protein 14-3-3 in the CSF was determined through ELISA immune assays in most of the cases. Positive results were taken according to each laboratory standard. The diagnostic criteria for probable cases were defined as follows:Citation1,Citation3 (1) a rapidly progressive dementia (<2 years), (2) abnormal EEG findings with triphasic morphology and/or the presence of 14-3-3 protein in cerebrospinal fluid examination and (3) at least two of the following 4 clinical signs: (a) myoclonus, (b) ataxia and or/visual signs and symptoms, (c) extrapyramidal or pyramidal signs and symptoms and (d) akinetic mutism. Patients with the clinical signs of CJD but without the classic EEG or CSF abnormalities (either not present or investigation not available) were classified as possible CJD. The definite diagnosis required a neuropathological examination or detection of scrapie prion protein by western blot analysis. Exclusion criteria included a positive thyroid test and antinuclear antibodies among other tests.
Conclusions
CJD is rarely recognized, however, the rapid development of signs and symptoms in addition to abnormalities in the MRI and EEG will aid the diagnosis in many cases despite not having timely access to CSF protein 14-3-3. Specially, the early identification of new onset of depression, agitation, irritability or memory loss should not be overlooked in middle age patients who do not have a previous history of a psychiatric or neurologic illness. Unfortunately, there is no treatment available, however, the recognition of more cases will allow us to understand the epidemiology and pathophysiology of this rare and devastating disease.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Figures and Tables
Figure 1 Bilateral hyperintensities of the basal ganglia in case 1 (A), 4 (B) and 7 (C), compared to a normal control (D).
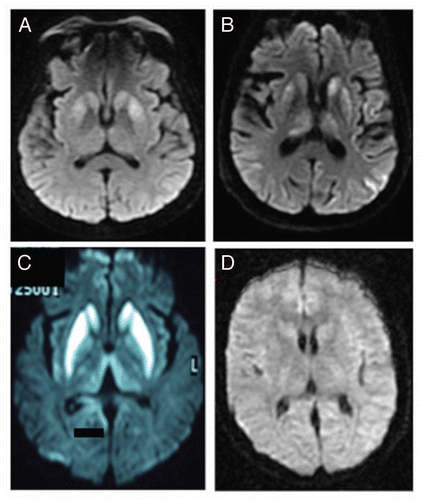
Figure 2 EEG of (A) Case4, (B) Case 5, (C) Case 6 and (D) control. In contrast to (D), figures (A–C) show triphasic waves (arrows) in a pseudoperiodic or periodic pattern.
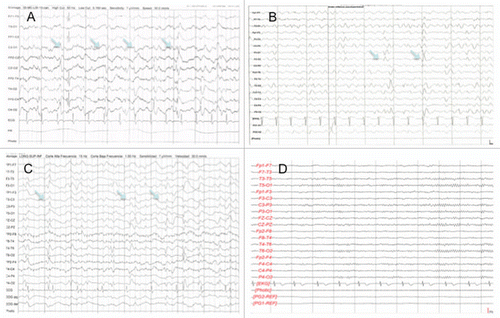
Table 1 Demographic, clinical and laboratory findings
Table 2 Mexican series of CJD
References
- World Health Organization. Manual for Surveillance of Human Transmissible Spongiform Encephalopathies Including Variant Creutzfeldt Jakob Disease 2003; Geneva World Health Organization
- Wleser HG, Schindler K, Zumsteg D. EEG in Creutzfeldt-Jakob disease. Clinical Neurophysiology 2006; 117:935 - 951
- Velasquez-Perez L, Rembao-Bojorquez J, Guevara J, Guadarrama-Torres RM, Trejo-Contreras A. Creutzfeldt-Jakob Disease in Mexico. Neuropathology 2007; 27:419 - 428
- Calderón-Garcidueñas AL, Sagastegui-Rodriguez JA, Canales-Ibarra C, Farías-García R. A case of Creutzfeldt-Jakob in the Mexican north-east and review of current concepts on prion disease. Gac Med Mex 2001; 137:589 - 594
- Martinez-Barros M, Ramos Peek J, Vega R, Escobar A. La enfermedad de Creutzfeldt-Jakob. Correlación clínica, electrofisiológica e histopatológica. Gac Med Mex 1996; 131:591 - 596
- Kltamoto T. Slow Virus Infection Research Comitte. Slow Virus Infection Research Committte. Clinical Stages of Creutzfeldt-Jakob disease. Annual report of the Slow Virus Infection Research Committee 1999; Tokyo The Ministry of Health and Welfare of Japan 9
- Schröter A, Zerr I, Henkel K, Tschampa HJ, Finkenstaedt M, Poser S. Magnetic resonance imaging in the clinical diagnosis of Creutzfeldt-Jakob Disease. Arch Neurol 2000; 57:1751 - 1757
- Shiga Y, Miyazawa K, Sato S, Fukushima R, Shibuya S, Sato Y, et al. Diffusion-weighted MRI abnormalities as an early diagnostic marker for Creutzfeldt-Jakob disease. Neurology 2004; 10:443 - 449
- Zeidler M, Green A. Advances in diagnosing Creutzfeldt-Jakob disease with MRI and CSF 14-3-3-protein analysis. Neurology 2004; 63:410 - 441
- Zerr I, Pocchiari M, Collins S, Brandel JP, de Pedro Cuesta J, Knight RS, et al. Analysis of EEG and CSF 14-3-3-proteins as aids to the diagnosis of Creutzfeldt-Jakob disease. Neurology 2000; 55:811 - 815
- Kellar JA, Lees VW. Risk management of the transmissible spongiform encephalopthies in North America. Rev Sci Tech 2003; 22:201 - 225