Abstract
The soluble cellular prion protein (PrPC) is best known for its association with prion disease (PrD) through its conversion to a pathogenic insoluble isoform (PrPSc). However, its deleterious effects independent of PrPSc have recently been observed not only in PrD but also in Alzheimer disease (AD), two diseases which mainly affect cognition. At the same time, PrPC itself seems to have broad physiologic functions including involvement in cognitive processes. The PrPC that is believed to be soluble and monomeric has so far been the only PrP conformer observed in the uninfected brain. In 2006, we identified an insoluble PrPC conformer (termed iPrPC) in uninfected human and animal brains. Remarkably, the PrPSc-like iPrPC shares the immunoreactivity behavior and fragmentation with a newly-identified PrPSc species in a novel human PrD termed variably protease-sensitive prionopathy. Moreover, iPrPC has been observed as the major PrP species that interacts with amyloid β (Aβ) in AD. This article highlights evidence of PrP involvement in two putatively beneficial and deleterious PrP-implicated pathways in cognition, and hypothesizes first, that beneficial and deleterious effects of PrPC are attributable to the chameleon-like conformation of the protein and second, that the iPrPC conformer is associated with PrD and AD.
Acknowledgments
The authors are grateful to Dr. Pedro Fernandez-Funez for helpful comments. This work was supported by the National Institutes of Health R01NS062787, the University Center on Aging and Health with the support of the McGregor Foundation and the President's Discretionary Fund (Case Western Reserve University), the Alliance BioSecure and the CJD Foundation.
Figures and Tables
Figure 1 Diagram of the pathway involved in prion- or Aβ-induced dysfunction of memory and cognition as well as cell death. PrPSc or Aβ oligomers impair memory and cognition, and induce cell death by binding to PrPC at the synapse but the downstream molecular events (the black box with a question mark) of the PrPSc-PrPC or PrPC-Aβ complex are unknown. The iPrPC species derived from the soluble PrPC might play a role in one or more of the following events: (1) long-term memory storage; (2) PrPSc formation in classic CJD; (3) initiating VPSPr; and (4) facilitating formation of Aβ42 fibrils in AD.
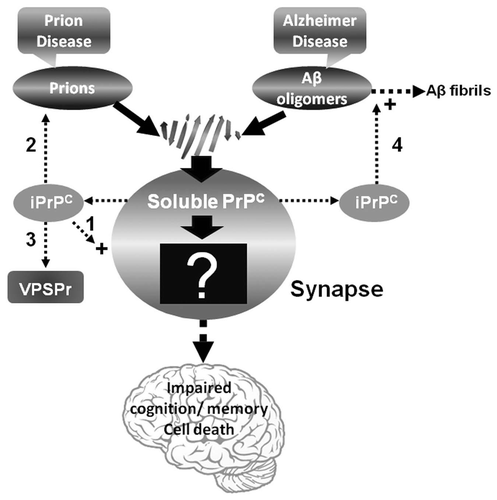
Figure 2 Comparison of PK-resistant PrP core fragments from non-CJD, VPSPr and sCJD. PrP captured with g5p from brain homogenates of three non-CJD subjects was treated with PK and PNGase F prior to SDS-PAGE and immunoblotting with anti-PrP antibody 1E4. Fifty µl of insoluble fraction (P2) equivalent to 500 µl of supernatant (S1) from a low-speed centrifuge was used for each non-CJD subject. In these highly concentrated samples from non-CJD subjects, three PK-resistant PrP (PrPres) fragments migrating at ∼20 kDa, ∼17–18 kDa and ∼6–7 kDa were detected with 1E4. But these PrPres fragments were not detectable by 3F4 (data not shown). In contrast, three PrP fragments with similar gel mobility were also detected in only 5 µl of S1 preparation from a VPSPr case (129MV) by 1E4 after treatment with PK and PNGase F. However, only one PrP band was detected in sCJD samples after PK and PNGase F treatment.